
Identification of Susceptibility Genes for Fusarium oxysporum in Cucumber via Comparative Proteomic Analysis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Foc Inoculation
2.2. Total Protein Extraction and Itraq Analysis
2.3. Identification of Proteins and Bioinformatic Analysis
2.4. RNA Isolation and Expression Pattern Analysis
2.5. Statistical Analysis
3. Results
3.1. Symptoms of Foc Infection on Rijiecheng and Superina Roots
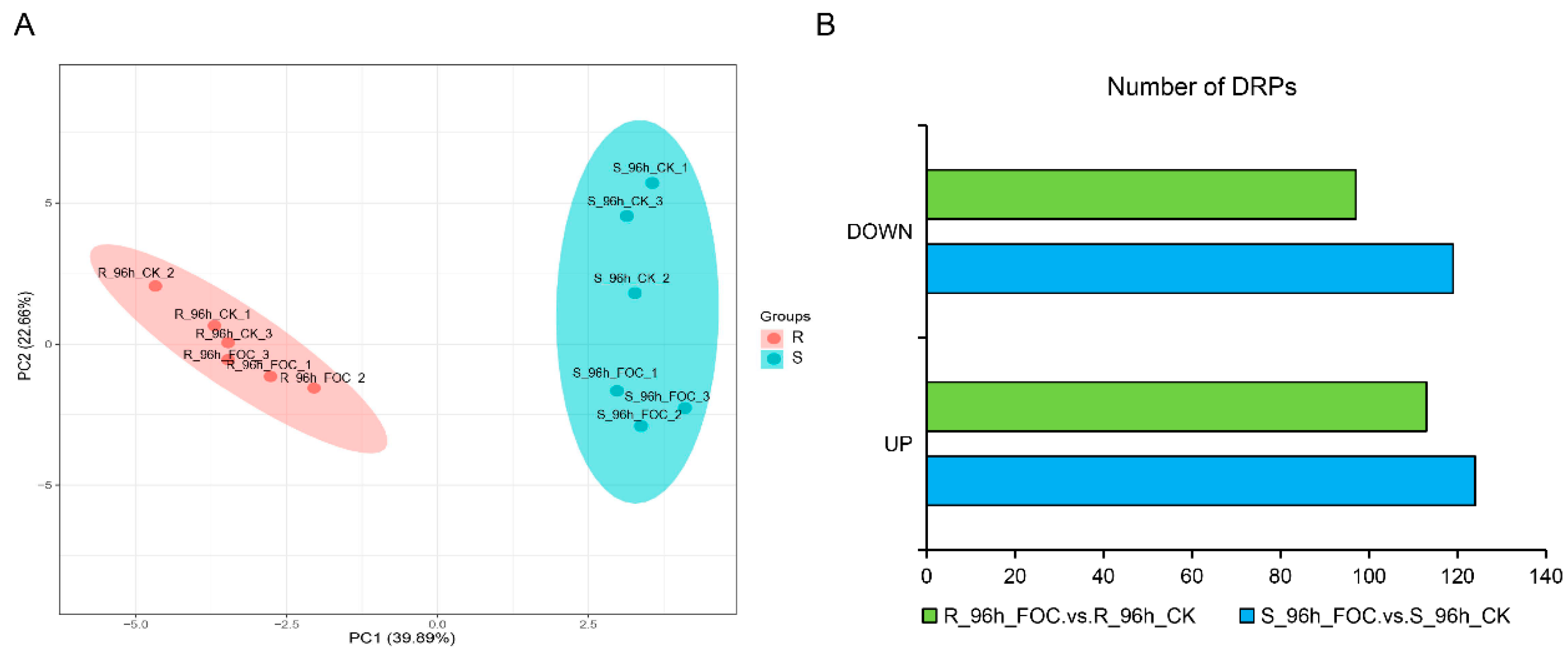
3.2. Identification of the DRPs in Rijiecheng and Superina Roots by iTRAQ
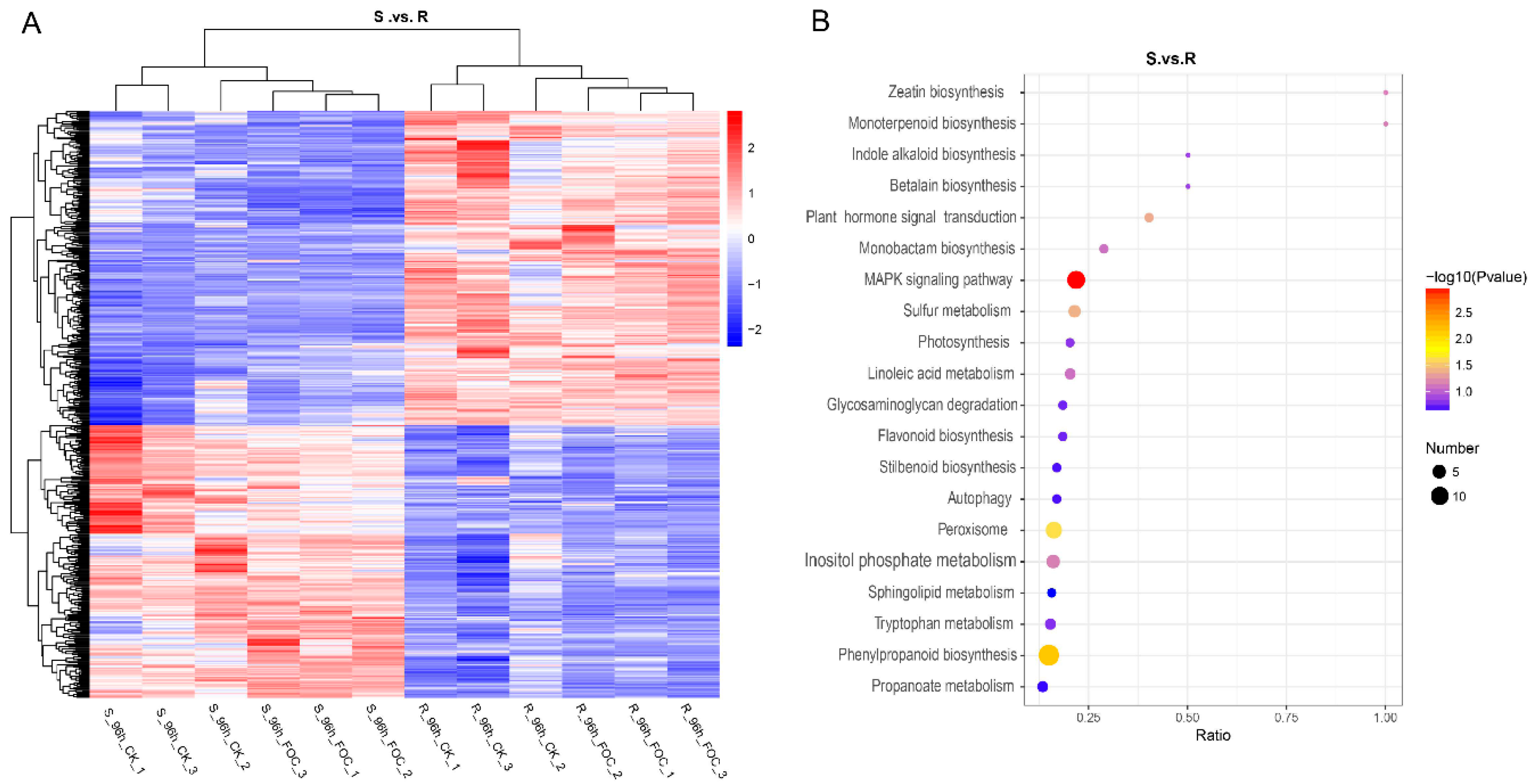
3.3. Comparing the Expression of Proteins between the Resistant and Susceptible Lines
3.4. Profile of Proteins Altered in Cucumber Inoculated with Foc
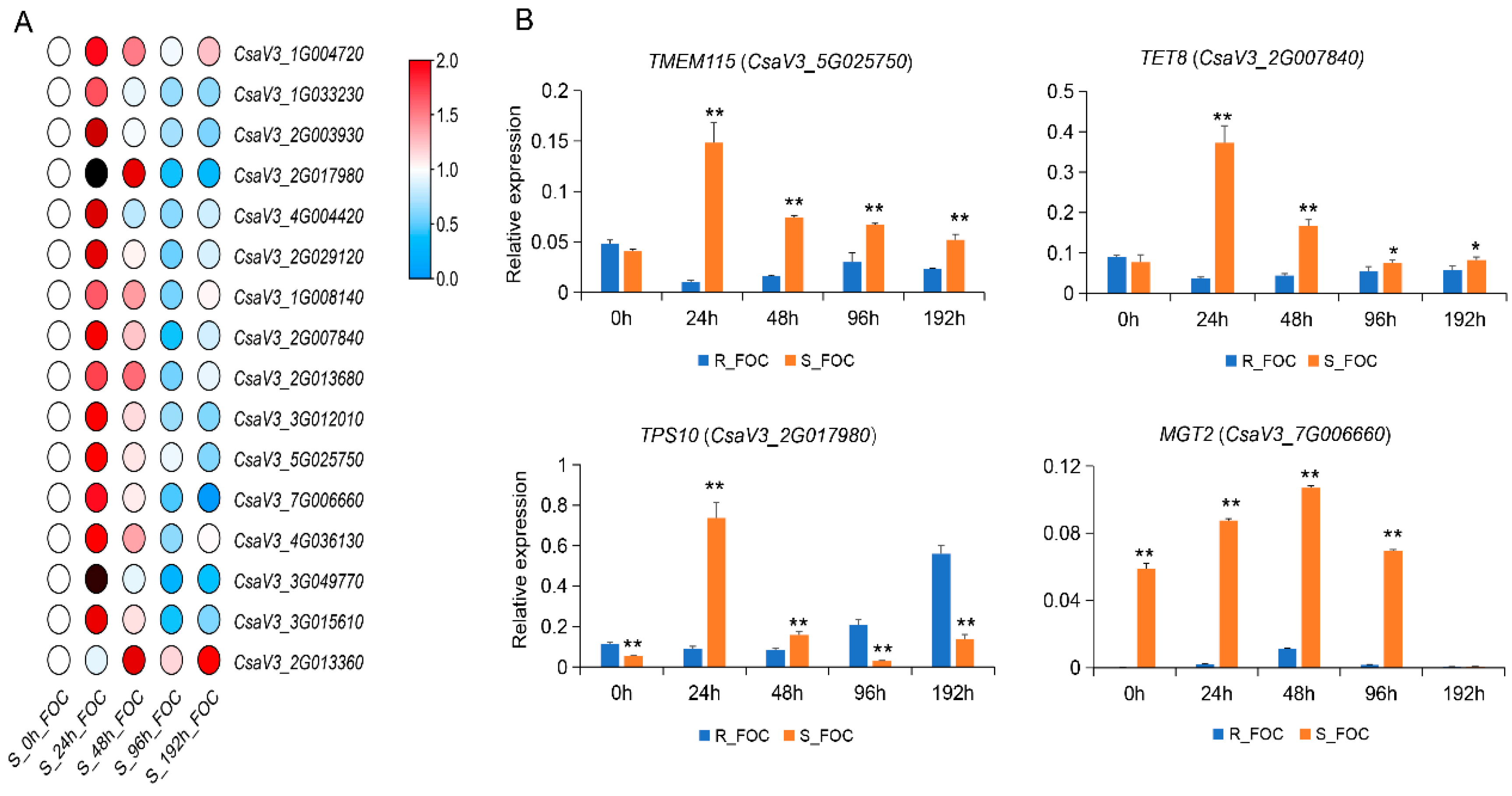
3.5. Validation of Genes Encoding DRPs by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Huang, S.; Li, R.; Zhang, Z.; Li, L.; Gu, X.; Fan, W.; Lucas, W.J.; Wang, X.; Xie, B.; Ni, P.; et al. The genome of the cucumber, Cucumis sativus L. Nat. Genet. 2009, 41, 1275–1281. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.-L.; Lin, X.-G.; Wang, J.-H.; Shen, W.-S.; Wu, S.; Peng, S.-P.; Mao, T.-T. Arbuscular Mycorrhizal Fungal Inoculation Enhances Suppression of Cucumber Fusarium Wilt in Greenhouse Soils. Pedosphere 2010, 20, 586–593. [Google Scholar] [CrossRef]
- Ahn, P.; Chung, H.-S.; Lee, Y.-H. Vegetative Compatibility Groups and Pathogenicity Among Isolates of Fusarium oxysporum f. sp. cucumerinum. Plant Dis. 1998, 82, 244–246. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Wu, F. Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiol. Ecol. 2012, 80, 469–478. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Li, Y.; Li, Y.; Wang, M.; Mur, L.A.J.; Shen, Q.; Guo, S. Nitrate mediated resistance against Fusarium infection in cucumber plants acts via photorespiration. Plant Cell Environ. 2021, 44, 3412–3431. [Google Scholar] [CrossRef]
- Dong, J.; Wang, Y.; Xian, Q.; Chen, X.; Xu, J. Transcriptome analysis reveals ethylene-mediated defense responses to Fusarium oxysporum f. sp. cucumerinum infection in Cucumis sativus L. BMC Plant Biol. 2020, 20, 1–10. [Google Scholar] [CrossRef]
- Du, N.; Shi, L.; Du, L.; Yuan, Y.; Li, B.; Sang, T.; Sun, J.; Shu, S.; Guo, S. Effect of vinegar residue compost amendments on cucumber growth and Fusarium wilt. Environ. Sci. Pollut. Res. 2015, 22, 19133–19141. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Aguilar, M.I.; Guirado, M.L.; Alvarez, A.; Gomez, J. First report of fusarium wilt of cucumber caused by Fusarium oxysporum in Spain. Plant Pathol. 2003, 52, 410. [Google Scholar] [CrossRef]
- Chen, L.-H.; Huang, X.-Q.; Zhang, F.-G.; Zhao, D.-K.; Yang, X.-M.; Shen, Q.-R. Application of Trichoderma harzianum SQR-T037 bio-organic fertiliser significantly controls Fusarium wilt and affects the microbial communities of continuously cropped soil of cucumber. J. Sci. Food Agric. 2012, 92, 2465–2470. [Google Scholar] [CrossRef]
- Huang, X.; Shi, D.; Sun, F.; Lu, H.; Liu, J.; Wu, W. Efficacy of sludge and manure compost amendments against Fusarium wilt of cucumber. Environ. Sci. Pollut. Res. 2012, 19, 3895–3905. [Google Scholar] [CrossRef] [PubMed]
- Keinath, A.P.; Hassell, R.L. Control of Fusarium Wilt of Watermelon by Grafting onto Bottlegourd or Interspecific Hybrid Squash Despite Colonization of Rootstocks by Fusarium. Plant Dis. 2014, 98, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Wang, M.; Sun, Y.; Gu, Z.; Wang, R.; Saydin, A.; Shen, Q.; Guo, S. Nitrate Increased Cucumber Tolerance to Fusarium Wilt by Regulating Fungal Toxin Production and Distribution. Toxins 2017, 9, 100. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Li, K.; Ma, Z.; Zou, H.; Jin, H.; Wang, J. Cucumber Fusarium wilt resistance induced by intercropping with celery differs from that induced by the cucumber genotype and is related to sulfur-containing allelochemicals. Sci. Hortic. 2020, 271, 109475. [Google Scholar] [CrossRef]
- Gu, Z.; Wang, M.; Wang, Y.; Zhu, L.; Mur, L.A.J.; Hu, J.; Guo, S. Nitrate Stabilizes the Rhizospheric Fungal Community to Suppress Fusarium Wilt Disease in Cucumber. Mol. Plant-Microbe Interact. 2020, 33, 590–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, L.K.; Wehner, T.C. Review of Genes and Linkage Groups in Cucumber. HortScience 1990, 25, 605–615. [Google Scholar] [CrossRef]
- Vakalounakis, D.J. Inheritance and genetic linkage of Fusarium wilt (Fusarium oxysporum f. sp. cucumerinum race 1) and scab (Cladosporium cucumerinum) resistance genes in cucumber (Cucumis sativus L.). Ann. Appl. Biol. 1993, 123, 359–365. [Google Scholar] [CrossRef]
- Liu, D.L.; Yang, R.H.; Ha, Y.H. Research on genetic characteristics of cucumbers resisting to Fusarium wilt. Tianjin Agric. Sci. 2003, 9, 33–35. [Google Scholar]
- Vakalounakis, D.J. Inheritance and linkage of resistance in cucumber line SMR-18 to races 1 and 2 of Fusarium oxysporum f. sp. cucumerium. Plant Pathol. 2015, 44, 169–172. [Google Scholar]
- Dong, J.P.; Qi, X.H.; Xu, Q.; Chen, X.H. Segregation and identification of Fusarium oxysporum f. sp. cucumerinum and analysis of cucumber varieties’ resistance difference. Mol. Plant Breed. 2017, 15, 3648–3653. [Google Scholar]
- Zaidi, S.S.E.A.; Mukhtar, S.; Mansoor, S. Genome Editing: Targeting Susceptibility Genes for Plant Disease Resistance. Trends Biotechnol. 2018, 36, 898–906. [Google Scholar] [CrossRef]
- Wang, Y.P.; Cheng, X.; Shan, Q.W.; Zhang, Y.; Liu, J.X.; Gao, C.X.; Qiu, J.L. Simultaneous editing of three homoeo-alleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechinol. 2013, 31, 686–688. [Google Scholar]
- Shan, Q.; Wang, Y.; Li, J.; Zhang, Y.; Chen, K.; Liang, Z.; Zhang, K.; Liu, J.; Xi, J.J.; Qiu, J.-L.; et al. Targeted genome modification of crop plants using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 686–688. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Garcia, J.; Spencer, D.; Thieron, H.; Reinstädler, A.; Hammond-Kosack, K.; Phillips, A.L.; Panstruga, R. mlo-based powdery mildew resistance in hexaploid bread wheat generated by a non-transgenic TILLING approach. Plant Biotechnol. J. 2016, 15, 367–378. [Google Scholar] [CrossRef]
- Kusch, S.; Panstruga, R. mlo-Based Resistance: An Apparently Universal “Weapon” to Defeat Powdery Mildew Disease. Mol. PlantMicrobe Interact. 2017, 30, 179–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.C.; Panstruga, R.; Elliott, C.; Müller, J.; Devoto, A.; Yoon, H.W.; Park, H.C.; Cho, M.J.; Schulze-Lefert, P. Calmodulin interacts with MLO protein to regulate defence against mildew in barley. Nature 2002, 416, 447–451. [Google Scholar] [CrossRef]
- Nekrasov, V.; Wang, C.; Win, J.; Lanz, C.; Weigel, D.; Kamoun, S. Rapid generation of a transgene-free powdery mildew resistant tomato by genome deletion. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, D.-Y.; Guo, Y.; Cheng, Y.; Hu, Y.; Xiao, S.; Wang, Y.; Wen, Y.-Q. CRISPR/Cas9-mediated mutagenesis of VvMLO3 results in enhanced resistance to powdery mildew in grapevine (Vitis vinifera). Hortic. Res. 2020, 7, 1–14. [Google Scholar] [CrossRef]
- Pessina, S.; Lenzi, L.; Perazzolli, M.; Campa, M.; Costa, L.D.; Urso, S.; Valè, G.; Salamini, F.; Velasco, R.; Malnoy, M. Knockdown of MLO genes reduces susceptibility to powdery mildew in grapevine. Hortic. Res. 2016, 3, 16016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, J.A.; Appiano, M.; Santillán Martínez, M.; Hermans, F.W.; Vriezen, W.H.; Visser, R.G.; Bai, Y.; Schouten, H.J. A transposable element insertion in the susceptibility gene CsaMLO8 results in hypocotyl resistance to powdery mildew in cucumber. BMC Plant Biol. 2015, 15, 243. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.W.; Bai, Y.; Wu, G.H.; Zou, S.H.; Chen, Y.F.; Gao, C.X.; Tang, D.Z. Simultaneous modification of three homoeologs of TaEDR1 by genome editing enhances powdery mildew resistance in wheat. Plant J. 2017, 91, 714–724. [Google Scholar] [CrossRef] [Green Version]
- Stover, E.D.; Driggers, R.; Richardson, M.L.; Hall, D.G.; Lee, R.F. Incidence and severity of Asiatic citrus canker on diverse citrus and citrus-related germplasm in a Florida field planting. Hortscience 2014, 49, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.H.; Chen, S.C.; Lei, T.G.; Xu, L.Z.; He, Y.R.; Wu, L.; Yao, L.X.; Zou, X.P. Engineering canker-resistant plants through CRISPR/Cas9-targeted editing of the susceptibility gene CsLOB1 promoter in citrus. Plant Biotechnol. J. 2017, 15, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Pyott, D.E.; Sheehan, E.; Molnar, A. Engineering of CRISPR/Cas9-mediatedpotyvirus resistance in transgene-free Arabidopsis plants. Mol. Plant Pathol. 2016, 17, 1276–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekaran, J.; Brumin, M.; Wolf, D.; Leibman, D.; Klap, C.; Pearlsman, M.; Sherman, A.; Arazi, T.; Gal-On, A. Development of broad virus resistance in non-transgenic cucumber using CRISPR/Cas9 technology. Mol. Plant Pathol. 2016, 17, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Liu, X.; Yan, Y.; Wang, W.; Gebretsadik, K.; Qi, X.; Xu, Q.; Chen, X. Comparative proteomic analysis of cucumber powdery mildew resistance between a single-segment substitution line and its recurrent parent. Hortic. Res. 2019, 6, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Noirel, J.; Evans, C.; Salim, M.; Mukherjee, J.; Ow, S.Y.; Pandhal, J.; Pham, T.K.; Biggs, C.A.; Wright, P.C. Methods in Quantitative Proteomics: Setting iTRAQ on the Right Track. Curr. Proteom. 2011, 8, 17–30. [Google Scholar] [CrossRef]
- Wang, X.; Zenda, T.; Liu, S.; Liu, G.; Jin, H.; Dai, L.; Dong, A.; Yang, Y.; Duan, H. Comparative Proteomics and Physiological Analyses Reveal Important Maize Filling-Kernel Drought-Responsive Genes and Metabolic Pathways. Int. J. Mol. Sci. 2019, 20, 3743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ana, C.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2016, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, H.; Goto, S.; Sato, K.; Fujibuchi, W.; Bono, H.; Kanehisa, M. KEGG: Kyotoencyclopedia of genes and genomes. Nucleic Acids Res. 1999, 27, 29–34. [Google Scholar] [CrossRef] [Green Version]
- Snyder, W.C.; Hansen, H.N. The species concept in Fusarium. Am. J. Bot. 1940, 27, 64–67. [Google Scholar] [CrossRef]
- Van Schie, C.C.; Takken, F.L. Susceptibility Genes 101: How to be a good host. Annu. Rev. Phytopathol. 2014, 52, 551–581. [Google Scholar] [CrossRef]
- Lapin, D.; van den Ackerveken, G. Susceptibility toplant disease: More than a failure of host immunity. Trends Plant Sci. 2013, 18, 546–554. [Google Scholar] [CrossRef]
- Parisi, C.; Tillie, P.; Rodríguez-Cerezo, E. The global pipeline of GM crops out to 2020. Nat. Biotechnol. 2016, 34, 31–36. [Google Scholar] [CrossRef]
- Yin, K.; Gao, C.; Qiu, J.-L. Progress and prospects in plant genome editing. Nat. Plants 2017, 3, 17107. [Google Scholar] [CrossRef] [PubMed]
- Bastet, A.; Robaglia, C.; Gallois, J.-L. eIF4E Resistance: Natural Variation Should Guide Gene Editing. Trends Plant Sci. 2017, 22, 411–419. [Google Scholar] [CrossRef]
- Chen, L.-Q.; Qu, X.-Q.; Hou, B.-H.; Sosso, D.; Osorio, S.; Fernie, A.R.; Frommer, W.B. Sucrose Efflux Mediated by SWEET Proteins as a Key Step for Phloem Transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Antony, G.; Zhou, J.; Huang, S.; Li, T.; Liu, B.; White, F.; Yang, B. Rice xa13 Recessive Resistance to Bacterial Blight Is Defeated by Induction of the Disease Susceptibility Gene Os-11N3. Plant Cell 2010, 22, 3864–3876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, W.; Zhou, H.; Bi, H.; Fromm, M.; Yang, B.; Weeks, D.P. Demonstration of CRISPR/Cas9/sgRNA mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 2013, 41, e188. [Google Scholar] [CrossRef]
- Li, T.; Liu, B.; Spalding, M.H.; Weeks, D.P.; Yang, B. High-efficiency TALEN-based gene editing produces disease-resistant rice. Nat. Biotechnol. 2012, 30, 390–392. [Google Scholar] [CrossRef]
- Garbutt, C.C.; Bangalore, P.V.; Kannar, P.; Mukhtar, M.S. Getting to the edge: Protein dynamical networks as a new frontier in plant-microbe interactions. Front. Plant Sci. 2014, 5, 312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naqvi, R.Z.; Zaidi, S.S.; Akhtar, K.P.; Strickler, S.; Woldemariam, M.; Mishra, B.; Mukhtar, M.S.; Scheffler, B.E.; Scheffler, J.A.; Jander, G.; et al. Transcriptomics reveals multiple resistance mechanisms against cotton leaf curl disease in a naturally immune cotton species, Gossypium arboreum. Sci. Rep. 2017, 7, 15880. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Le Novère, N. Quantitative and logic modelling of molecular and gene networks. Nat. Rev. Genet. 2015, 16, 146–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Zhao, D.; Dong, J.; Kong, X.; Zhang, Q.; Li, T.; Meng, Y.; Shan, W. AtRTP5 negatively regulates plant resistance to Phytophthora pathogens by modulating the biosynthesis of endogenous jasmonic acid and salicylic acid. Mol. Plant. Pathol. 2019, 21, 95–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, T.; Mao, X.; Zhang, H.; Zhang, Y.; Fu, M.; Sun, Z.; Kuai, P.; Lou, Y.; Fang, Y. Lamin-like Proteins Negatively Regulate Plant Immunity through NAC with transmembrane MOTIF1-LIKE9 and nonexpressor of PR GENES1 in Arabidopsis thaliana. Mol. Plant. 2017, 10, 1334–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, E.C.; Chen, Y.C.; Mudgett, M.B.; Sattely, E.S. Arabidopsis UGT76B1 glycosylates N-hydroxy-pipecolic acid and inactivates systemic acquired resistance in tomato. Plant Cell 2021, 33, 750–765. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Wang, K.; Xian, Q.; Zhang, N.; Dong, J.; Chen, X. Identification of Susceptibility Genes for Fusarium oxysporum in Cucumber via Comparative Proteomic Analysis. Genes 2021, 12, 1781. https://doi.org/10.3390/genes12111781
Xu J, Wang K, Xian Q, Zhang N, Dong J, Chen X. Identification of Susceptibility Genes for Fusarium oxysporum in Cucumber via Comparative Proteomic Analysis. Genes. 2021; 12(11):1781. https://doi.org/10.3390/genes12111781
Chicago/Turabian StyleXu, Jun, Ke Wang, Qianqian Xian, Ningyuan Zhang, Jingping Dong, and Xuehao Chen. 2021. "Identification of Susceptibility Genes for Fusarium oxysporum in Cucumber via Comparative Proteomic Analysis" Genes 12, no. 11: 1781. https://doi.org/10.3390/genes12111781
APA StyleXu, J., Wang, K., Xian, Q., Zhang, N., Dong, J., & Chen, X. (2021). Identification of Susceptibility Genes for Fusarium oxysporum in Cucumber via Comparative Proteomic Analysis. Genes, 12(11), 1781. https://doi.org/10.3390/genes12111781