
Integrated Analysis Reveals a lncRNA–miRNA–mRNA Network Associated with Pigeon Skeletal Muscle Development

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Preparation
2.2. LncRNA Library Construction and Sequencing
2.3. miRNA Library Construction and Sequencing
2.4. LncRNA and mRNA Identification
2.5. miRNA Identification
2.6. Differentially Expression Analysis
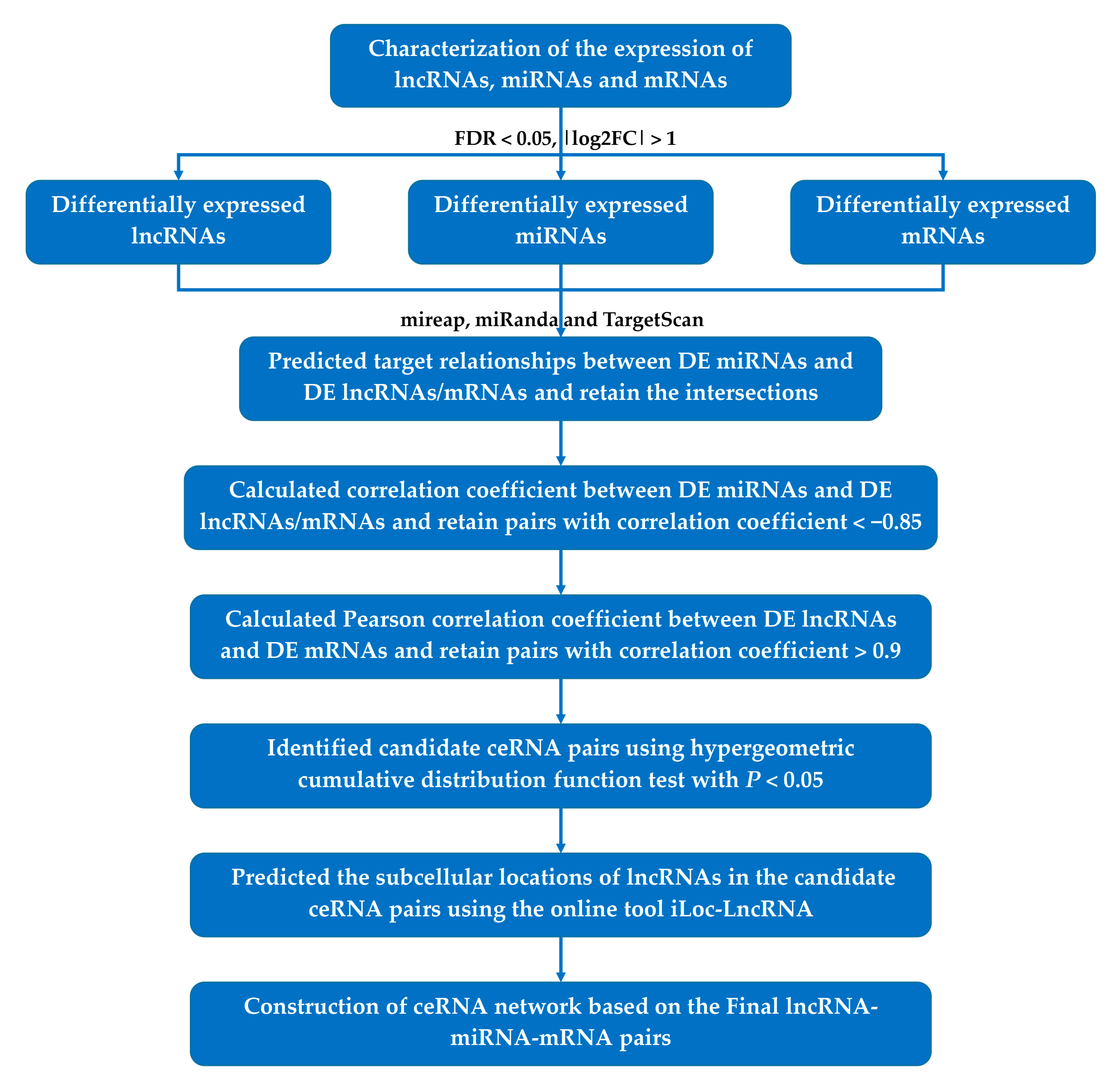
2.7. ceRNA Network Construction
2.8. Functional Enrichment Analysis of the ceRNA Network
2.9. Identification of Hub lncRNA–miRNA–mRNA Interactions
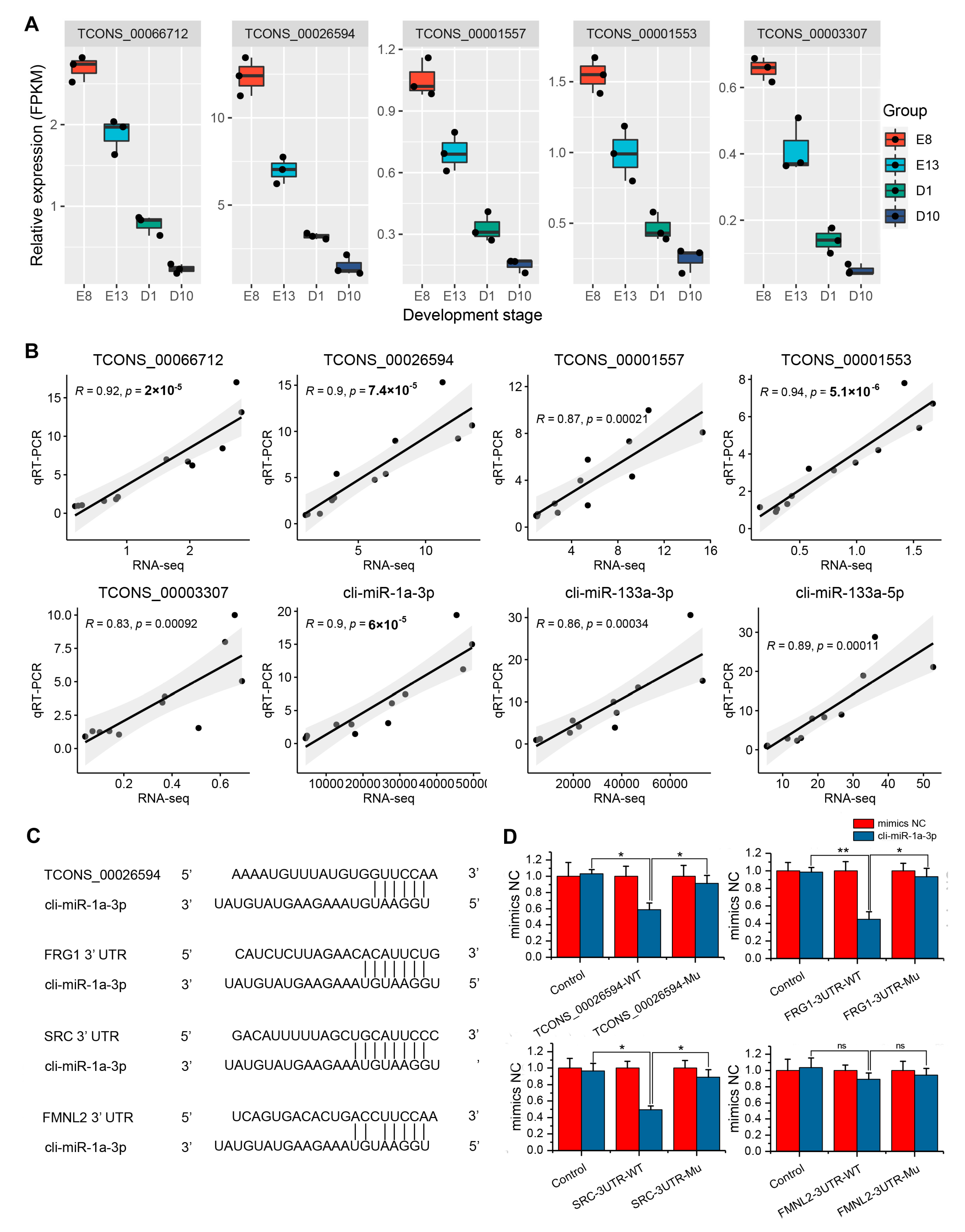
2.10. Validation of the Expression of ceRNA Interactions Using qRT-PCR
2.11. Dual-Luciferase Activity Assay
3. Results
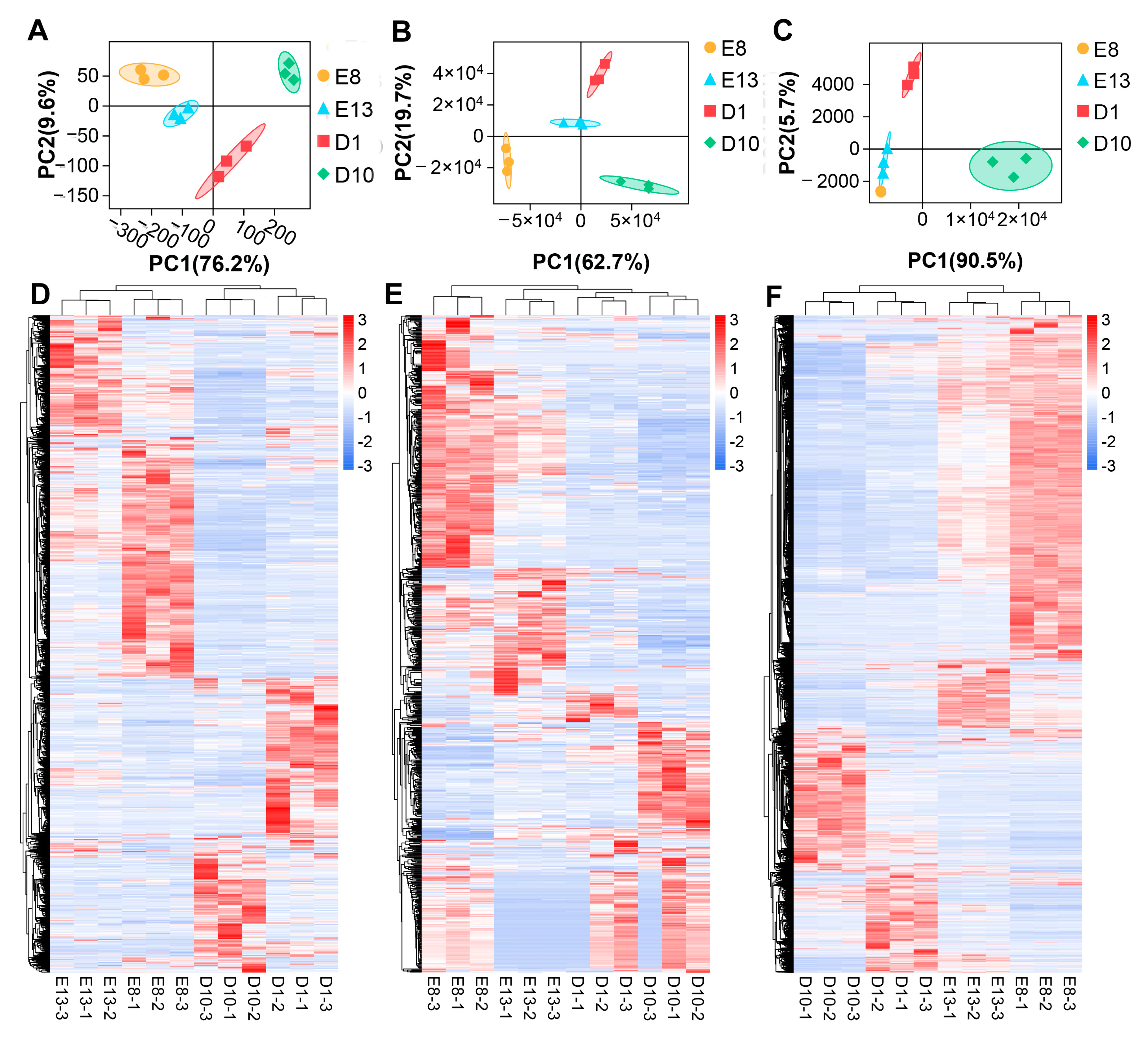
3.1. Identification of DE lncRNAs, miRNAs, and mRNAs
3.2. Construction of lncRNA-Associated ceRNA Network
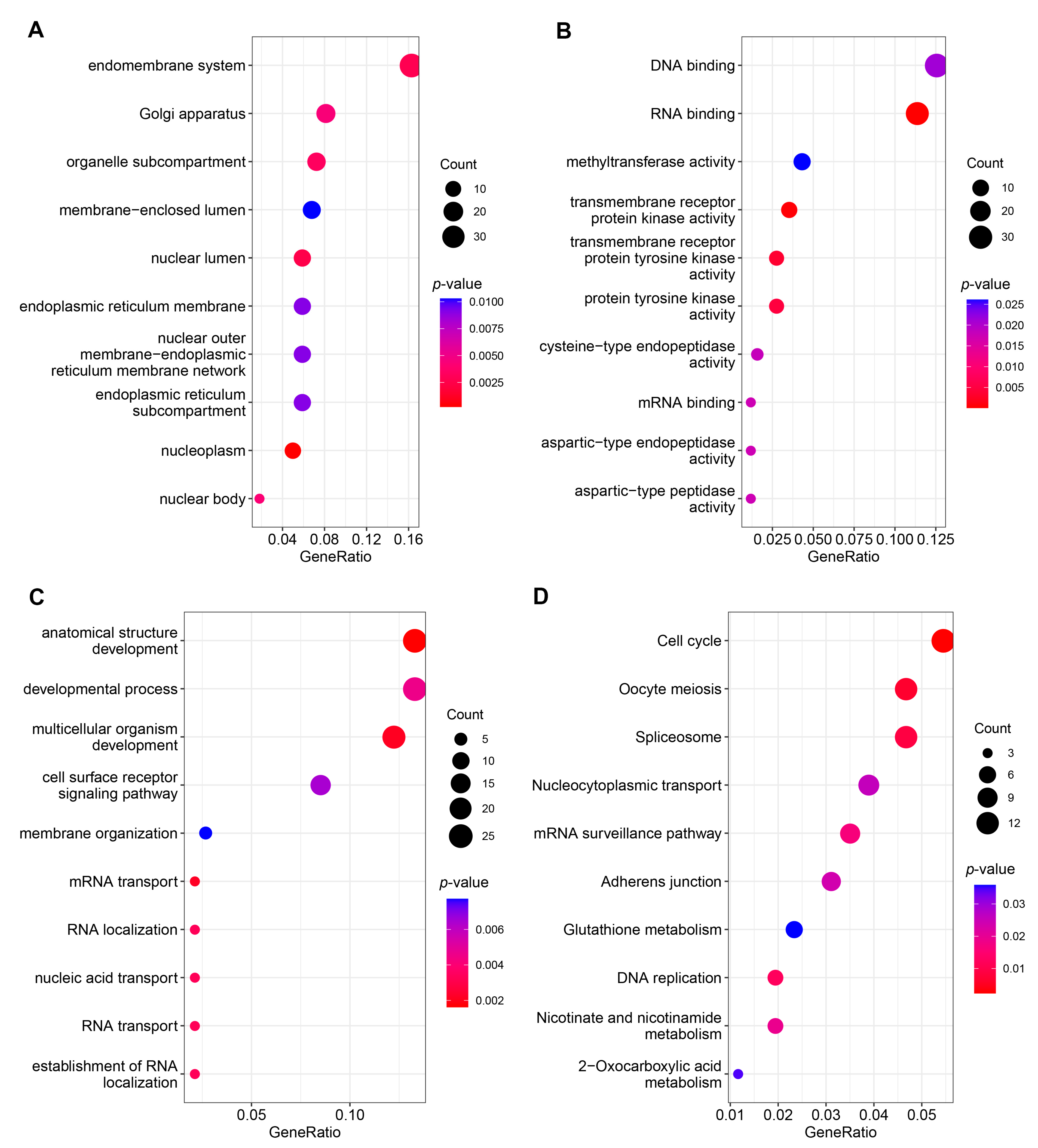
3.3. Functional Analysis of the ceRNA Network
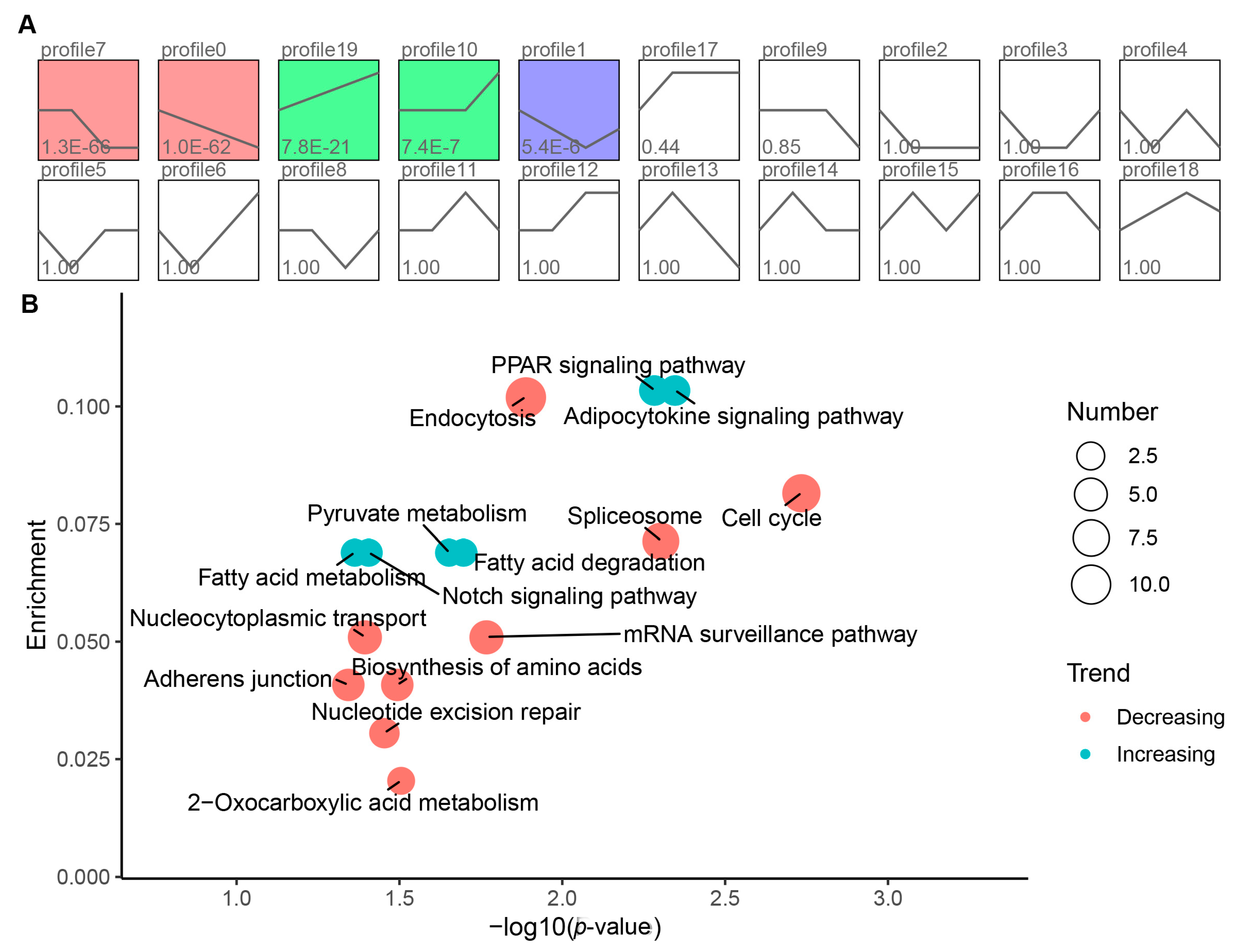
3.4. Short Time-Series Expression Miner and Function Analysis
3.5. Identification of Crucial lncRNA–miRNA–mRNA Interactions
3.6. Validation of ceRNA Interactions Using qRT-PCR and Dual-Luciferase Assay
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Petracci, M.; Cavani, C. Muscle growth and poultry meat quality issues. Nutrients 2012, 4, 1–12. [Google Scholar] [CrossRef]
- Machado Junior, P.C.; Chung, C.; Hagerman, A. Modeling Salmonella Spread in Broiler Production: Identifying Determinants and Control Strategies. Front. Vet. Sci. 2020, 7, 564. [Google Scholar] [CrossRef]
- Hasted, T.-L.; Sharif, S.; Boerlin, P.; Diarra, M.S. Immunostimulatory Potential of Fruits and Their Extracts in Poultry. Front. Immunol. 2021, 12, 641696. [Google Scholar] [CrossRef]
- Ji, F.; Zhang, D.; Shao, Y.; Yu, X.; Liu, X.; Shan, D.; Wang, Z. Changes in the diversity and composition of gut microbiota in pigeon squabs infected with Trichomonas gallinae. Sci. Rep. 2020, 10, 19978. [Google Scholar] [CrossRef]
- Pomianowski, J.F.; Mikulski, D.; Pudyszak, K.; Cooper, R.G.; Angowski, M.; Jóźwik, A.; Horbańczuk, J.O. Chemical composition, cholesterol content, and fatty acid profile of pigeon meat as influenced by meat-type breeds. Poult. Sci. 2009, 88, 1306–1309. [Google Scholar] [CrossRef]
- Ye, M.; Xu, M.; Chen, C.; He, Y.; Ding, M.; Ding, X.; Wei, W.; Yang, S.; Zhou, B. Expression analyses of candidate genes related to meat quality traits in squabs from two breeds of meat-type pigeon. J. Anim. Physiol. Anim. Nutr. 2018, 102, 727–735. [Google Scholar] [CrossRef]
- Yin, H.; He, H.; Cao, X.; Shen, X.; Han, S.; Cui, C.; Zhao, J.; Wei, Y.; Chen, Y.; Xia, L.; et al. MiR-148a-3p Regulates Skeletal Muscle Satellite Cell Differentiation and Apoptosis via the PI3K/AKT Signaling Pathway by Targeting Meox2. Front. Genet. 2020, 11, 512. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Xu, C.; Guo, J.; Liu, K.; Hu, Y.; Wu, D.; Fang, H.; Zou, Y.; Wei, Z.; Wang, Z.; et al. Cis- and Trans-Acting Expression Quantitative Trait Loci of Long Non-Coding RNA in 2,549 Cancers With Potential Clinical and Therapeutic Implications. Front. Oncol. 2020, 10, 602104. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, S.; Yan, Y.; Li, S.; Tong, H. SPARCL1 Influences Bovine Skeletal Muscle-Derived Satellite Cell Migration and Differentiation through an ITGB1-Mediated Signaling Pathway. Animals 2020, 10, 1361. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Huang, W.; Chen, B.; Jebessa Bekele, E.; Chen, X.; Cai, B.; Nie, Q. gga-mir-133a-3p Regulates Myoblasts Proliferation and Differentiation by Targeting PRRX1. Front. Genet. 2018, 9, 577. [Google Scholar] [CrossRef]
- Yu, J.-A.; Wang, Z.; Yang, X.; Ma, M.; Li, Z.; Nie, Q. LncRNA-FKBP1C regulates muscle fiber type switching by affecting the stability of MYH1B. Cell Death Discov. 2021, 7, 73. [Google Scholar] [CrossRef] [PubMed]
- Cai, B.; Li, Z.; Ma, M.; Wang, Z.; Han, P.; Abdalla, B.A.; Nie, Q.; Zhang, X. LncRNA-Six1 Encodes a Micropeptide to Activate Six1 in Cis and Is Involved in Cell Proliferation and Muscle Growth. Front. Physiol. 2017, 8, 230. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Abdalla, B.A.; Zheng, M.; He, X.; Cai, B.; Han, P.; Ouyang, H.; Chen, B.; Nie, Q.; Zhang, X. Systematic transcriptome-wide analysis of mRNA–miRNA interactions reveals the involvement of miR-142-5p and its target (FOXO3) in skeletal muscle growth in chickens. Mol. Genet. Genom. 2018, 293, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, S.W.; Han, J.S.; Shin, S.P.; Lee, S.I.; Park, T.S. Functional analyses of miRNA-146b-5p during myogenic proliferation and differentiation in chicken myoblasts. BMC Mol. Cell Biol. 2020, 21, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Chen, F.; Wu, P.; Li, T.; He, M.; Yin, X.; Shi, H.; Duan, Y.; Zhang, T.; Wang, J.; et al. MicroRNA-7 Targets the KLF4 Gene to Regulate the Proliferation and Differentiation of Chicken Primary Myoblasts. Front. Genet. 2020, 11, 842. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Gu, T.; Lu, L.; Cao, Z.; Song, Q.; Wang, Z.; Zhang, Y.; Chang, G.; Xu, Q.; Chen, G. Roles of miRNA-1 and miRNA-133 in the proliferation and differentiation of myoblasts in duck skeletal muscle. J. Cell. Physiol. 2019, 234, 3490–3499. [Google Scholar] [CrossRef] [PubMed]
- Salmena, L.; Poliseno, L.; Tay, Y.; Kats, L.; Pandolfi, P.P. A ceRNA hypothesis: The Rosetta Stone of a hidden RNA language? Cell 2011, 146, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Han, R.; Han, L.; Wang, S.; Li, H. Whole Transcriptome Analysis of Mesenchyme Tissue in Sika Deer Antler Revealed the CeRNAs Regulatory Network Associated With Antler Development. Front. Genet. 2020, 10, 1403. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Huang, K.; Wang, P.; Feng, T.; Shi, D.; Cui, K.; Luo, C.; Shafique, L.; Qian, Q.; Ruan, J.; et al. Comparison of Long Non-Coding RNA Expression Profiles of Cattle and Buffalo Differing in Muscle Characteristics. Front. Genet. 2020, 11, 98. [Google Scholar] [CrossRef]
- Yue, B.; Li, H.; Liu, M.; Wu, J.; Li, M.; Lei, C.; Huang, B.; Chen, H. Characterization of lncRNA–miRNA–mRNA Network to Reveal Potential Functional ceRNAs in Bovine Skeletal Muscle. Front. Genet. 2019, 10, 91. [Google Scholar] [CrossRef]
- Hong, L.; Gu, T.; He, Y.; Zhou, C.; Hu, Q.; Wang, X.; Zheng, E.; Huang, S.; Xu, Z.; Yang, J.; et al. Genome-Wide Analysis of Circular RNAs Mediated ceRNA Regulation in Porcine Embryonic Muscle Development. Front. Cell Dev. Biol. 2019, 7, 289. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Ren, Q.; Hua, L.; Chen, J.; Zhang, J.; Bai, H.; Li, H.; Xu, B.; Shi, Z.; Cao, H.; et al. Comprehensive Analysis of Differentially Expressed mRNA, lncRNA and circRNA and Their ceRNA Networks in the Longissimus Dorsi Muscle of Two Different Pig Breeds. Int. J. Mol. Sci. 2019, 20, 1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, M.; Cai, B.; Jiang, L.; Abdalla, B.A.; Li, Z.; Nie, Q.; Zhang, X. lncRNA-Six1 Is a Target of miR-1611 That Functions as a ceRNA to Regulate Six1 Protein Expression and Fiber Type Switching in Chicken Myogenesis. Cells 2018, 7, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, X.; Li, L.; Shi, G.; Chen, L.; Fang, C.; Li, M.; Li, C. MEG3 Promotes Differentiation of Porcine Satellite Cells by Sponging miR-423-5p to Relieve Inhibiting Effect on SRF. Cells 2020, 9, 449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Li, B.; Peng, W.; Ma, Y.; Huang, Y.; Lan, X.; Lei, C.; Qi, X.; Liu, G.E.; Chen, H. LncRNA-MEG3 promotes bovine myoblast differentiation by sponging miR-135. J. Cell. Physiol. 2019, 234, 18361–18370. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Zhang, T.; Zhang, S.; Huang, J.; Zhang, G.; Xie, K.; Wang, J.; Wu, H.; Dai, G. Identification of Long Non-Coding RNA-Associated Competing Endogenous RNA Network in the Differentiation of Chicken Preadipocytes. Genes 2019, 10, 795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.; Wang, X. miRDB: An online resource for microRNA target prediction and functional annotations. Nucleic Acids Res. 2014, 43, D146–D152. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. eLife 2015, 4, e05005. [Google Scholar] [CrossRef]
- Chen, L.-L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef]
- Zhao, J.; Pu, J.; Hao, B.; Huang, L.; Chen, J.; Hong, W.; Zhou, Y.; Li, B.; Ran, P. LncRNA RP11-86H7.1 promotes airway inflammation induced by TRAPM2.5 by acting as a ceRNA of miRNA-9-5p to regulate NFKB1 in HBECS. Sci. Rep. 2020, 10, 11587. [Google Scholar] [CrossRef]
- Su, Z.-D.; Huang, Y.; Zhang, Z.-Y.; Zhao, Y.-W.; Wang, D.; Chen, W.; Chou, K.-C.; Lin, H. iLoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L.; et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [CrossRef]
- Gao, S.; Wu, Z.; Feng, X.; Kajigaya, S.; Wang, X.; Young, N.S. Comprehensive network modeling from single cell RNA sequencing of human and mouse reveals well conserved transcription regulation of hematopoiesis. BMC Genom. 2020, 21, 849. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Neguembor, M.V.; Xynos, A.; Onorati, M.C.; Caccia, R.; Bortolanza, S.; Godio, C.; Pistoni, M.; Corona, D.F.; Schotta, G.; Gabellini, D. FSHD muscular dystrophy region gene 1 binds Suv4-20h1 histone methyltransferase and impairs myogenesis. J. Mol. Cell Biol. 2013, 5, 294–307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niu, A.; Wen, Y.; Liu, H.; Zhan, M.; Jin, B.; Li, Y.-P. Src mediates the mechanical activation of myogenesis by activating TNFα-converting enzyme. J. Cell Sci. 2013, 126, 4349–4357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wang, Z.; Tong, H.; Li, S.; Yan, Y. TCP11L2 promotes bovine skeletal muscle-derived satellite cell migration and differentiation via FMNL2. J. Cell. Physiol. 2020, 235, 7183–7193. [Google Scholar] [CrossRef]
- Horak, M.; Novak, J.; Bienertova-Vasku, J. Muscle-specific microRNAs in skeletal muscle development. Dev. Biol. 2016, 410, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chen, Y.; Jin, W.; Fu, S.; Li, D.; Zhang, Y.; Sun, G.; Jiang, R.; Han, R.; Li, Z.; et al. Analyses of MicroRNA and mRNA Expression Profiles Reveal the Crucial Interaction Networks and Pathways for Regulation of Chicken Breast Muscle Development. Front. Genet. 2019, 10, 197. [Google Scholar] [CrossRef] [Green Version]
- Collins, A.A.; Zou, K.; Li, Z.; Ying, S. Mechanism and Functions of Identified miRNAs in Poultry Skeletal Muscle Development —A Review. Ann. Anim. Sci. 2019, 19, 887–904. [Google Scholar] [CrossRef] [Green Version]
- Fan, H.; Ge, Y.; Ma, X.; Li, Z.; Shi, L.; Lin, L.; Xiao, J.; Chen, W.; Ni, P.; Yang, L.; et al. Long non-coding RNA CCDC144NL-AS1 sponges miR-143-3p and regulates MAP3K7 by acting as a competing endogenous RNA in gastric cancer. Cell Death Dis. 2020, 11, 521. [Google Scholar] [CrossRef]
- Shi, T.; Hu, W.; Hou, H.; Zhao, Z.; Shang, M.; Zhang, L. Identification and Comparative Analysis of Long Non-Coding RNA in the Skeletal Muscle of Two Dezhou Donkey Strains. Genes 2020, 11, 508. [Google Scholar] [CrossRef]
- Sun, J.; Xie, M.; Huang, Z.; Li, H.; Chen, T.; Sun, R.; Wang, J.; Xi, Q.; Wu, T.; Zhang, Y. Integrated analysis of non-coding RNA and mRNA expression profiles of 2 pig breeds differing in muscle traits1,2. J. Anim. Sci. 2017, 95, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Wang, S.; Wang, L.; Zhang, W.; Chen, W.; Lv, X.; Li, Y.; Hussain, Z.; Sun, W. Long Noncoding RNA (lncRNA) CTTN-IT1 Elevates Skeletal Muscle Satellite Cell Proliferation and Differentiation by Acting as ceRNA for YAP1 Through Absorbing miR-29a in Hu Sheep. Front. Genet. 2020, 11, 843. [Google Scholar] [CrossRef]
- Du, J.; Zhang, P.; Zhao, X.; He, J.; Xu, Y.; Zou, Q.; Luo, J.; Shen, L.; Gu, H.; Tang, Q.; et al. MicroRNA-351-5p mediates skeletal myogenesis by directly targeting lactamase-β and is regulated by lnc-mg. FASEB J. 2019, 33, 1911–1926. [Google Scholar] [CrossRef] [PubMed]
- Velleman, S.G. Relationship of Skeletal Muscle Development and Growth to Breast Muscle Myopathies: A Review. Avian Dis. 2015, 59, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Ji, Z.; Xuan, R.; Zhao, X.; Hou, L.; Li, Q.; Chu, Y.; Chao, T.; Wang, J. Differentially Expressed MiRNAs of Goat Submandibular Glands Among Three Developmental Stages Are Involved in Immune Functions. Front. Genet. 2021, 12, 678194. [Google Scholar] [CrossRef]
- Zhou, X.; Yuan, Q.; Zhang, C.; Dai, Z.; Du, C.; Wang, H.; Li, X.; Yang, S.; Zhao, A. Inhibition of Japanese encephalitis virus proliferation by long non-coding RNA SUSAJ1 in PK-15 cells. Virol. J. 2021, 18, 29. [Google Scholar] [CrossRef]
- Nakano, S.-i.; Nakamura, K.; Teramoto, N.; Yamanouchi, K.; Nishihara, M. Basic fibroblast growth factor is pro-adipogenic in rat skeletal muscle progenitor clone, 2G11 cells. Anim. Sci. J. 2016, 87, 99–108. [Google Scholar] [CrossRef]
- Zhu, M.H.; Chen, G.; Yang, Y.; Yang, J.T.; Qin, B.G.; Gu, L.Q. miR-217-5p regulates myogenesis in skeletal muscle stem cells by targeting FGFR2. Mol. Med. Rep. 2020, 22, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Goljanek-Whysall, K.; Pais, H.; Rathjen, T.; Sweetman, D.; Dalmay, T.; Münsterberg, A. Regulation of multiple target genes by miR-1 and miR-206 is pivotal for C2C12 myoblast differentiation. J. Cell Sci. 2012, 125, 3590–3600. [Google Scholar] [CrossRef] [Green Version]
- D’Alonzo, D.; Emch, F.H.; Shen, X.L.; Bruder, E.; De Geyter, C.; Zhang, H. Hectd1 is essential for embryogenesis in mice. Gene Expr. Patterns 2019, 34, 119064. [Google Scholar] [CrossRef]
- McFarland, D.C. Influence of growth factors on poultry myogenic satellite cells. Poult. Sci. 1999, 78, 747–758. [Google Scholar] [CrossRef]
- Lee, E.J.; Lee, H.J.; Kamli, M.R.; Pokharel, S.; Bhat, A.R.; Lee, Y.-H.; Choi, B.-H.; Chun, T.; Kang, S.W.; Lee, Y.S.; et al. Depot-specific gene expression profiles during differentiation and transdifferentiation of bovine muscle satellite cells, and differentiation of preadipocytes. Genomics 2012, 100, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Ahmad, S.; Jan, A.T.; Baig, M.H.; Lee, E.J.; Choi, I. Matrix gla protein: An extracellular matrix protein regulates myostatin expression in the muscle developmental program. Life Sci. 2017, 172, 55–63. [Google Scholar] [CrossRef]
- Tarroni, P.; Rossi, D.; Conti, A.; Sorrentino, V. Expression of the ryanodine receptor type 3 calcium release channel during development and differentiation of mammalian skeletal muscle cells. J. Biol. Chem. 1997, 272, 19808–19813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keren, A.; Tamir, Y.; Bengal, E. The p38 MAPK signaling pathway: A major regulator of skeletal muscle development. Mol. Cell. Endocrinol. 2006, 252, 224–230. [Google Scholar] [CrossRef]
- Pawlikowska, P.; Orzechowski, A. Role of insulin in myogenesis and mitachondrial respiration in relation to the ageing of skeletal muscles. Med. Weter.-Vet. Med.-Sci. Pract. 2006, 62, 380–384. [Google Scholar]
- Zheng, J.-N.; Li, Y.; Yan, Y.-M.; Shi, H.; Zou, T.-T.; Shao, W.-Q.; Wang, Q. Identification and Validation of Key Genes Associated With Systemic Sclerosis-Related Pulmonary Hypertension. Front. Genet. 2020, 11, 816. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Li, M.; He, J.; Huang, Z. Regulation of skeletal myogenesis by microRNAs. J. Cell. Physiol. 2020, 235, 87–104. [Google Scholar] [CrossRef] [PubMed]
- Hanel, M.L.; Wuebbles, R.D.; Jones, P.L. Muscular dystrophy candidate gene FRG1 is critical for muscle development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2009, 238, 1502. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Chen, C.; Han, S.; Chen, L.; Ding, H.; Lin, Y.; Zhang, G.; Xie, K.; Wang, J.; Dai, G. Integrated Analysis Reveals a lncRNA–miRNA–mRNA Network Associated with Pigeon Skeletal Muscle Development. Genes 2021, 12, 1787. https://doi.org/10.3390/genes12111787
Zhang T, Chen C, Han S, Chen L, Ding H, Lin Y, Zhang G, Xie K, Wang J, Dai G. Integrated Analysis Reveals a lncRNA–miRNA–mRNA Network Associated with Pigeon Skeletal Muscle Development. Genes. 2021; 12(11):1787. https://doi.org/10.3390/genes12111787
Chicago/Turabian StyleZhang, Tao, Can Chen, Shushu Han, Lan Chen, Hao Ding, Yueyue Lin, Genxi Zhang, Kaizhou Xie, Jinyu Wang, and Guojun Dai. 2021. "Integrated Analysis Reveals a lncRNA–miRNA–mRNA Network Associated with Pigeon Skeletal Muscle Development" Genes 12, no. 11: 1787. https://doi.org/10.3390/genes12111787
APA StyleZhang, T., Chen, C., Han, S., Chen, L., Ding, H., Lin, Y., Zhang, G., Xie, K., Wang, J., & Dai, G. (2021). Integrated Analysis Reveals a lncRNA–miRNA–mRNA Network Associated with Pigeon Skeletal Muscle Development. Genes, 12(11), 1787. https://doi.org/10.3390/genes12111787