
Genetic and Phenotypic Landscape of PRPH2-Associated Retinal Dystrophy in Japan

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Examinations
2.2. Genetic Screening
2.3. Statistical Analysis
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Broadgate, S.; Yu, J.; Downes, S.M.; Halford, S. Unravelling the genetics of inherited retinal dystrophies: Past, present and future. Prog. Retin. Eye Res. 2017, 59, 53–96. [Google Scholar] [CrossRef]
- Ku, C.A.; Pennesi, M.E. The new landscape of retinal gene therapy. Am. J. Med Genet. Part C Semin. Med Genet. 2020, 184, 846–859. [Google Scholar] [CrossRef]
- Boon, C.J.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog. Retin. Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog. Retin. Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, A.O.; Al Rashaed, S.; Neuhaus, C.; Bergmann, C.; Bolz, H.J. Peripherin mutations cause a distinct form of recessive Leber congenital amaurosis and dominant phenotypes in asymptomatic parents heterozygous for the mutation. Br. J. Ophthalmol. 2016, 100, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; Hahn, L.B.; Kajiwara, K.; Berson, E.L. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Invest. Ophthalmol. Vis. Sci. 1997, 38, 1972–1982. [Google Scholar] [PubMed]
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef] [Green Version]
- Donato, L.; Abdalla, E.M.; Scimone, C.; Alibrandi, S.; Rinaldi, C.; Nabil, K.M.; D′Angelo, R.; Sidoti, A. Impairments of Photoreceptor Outer Segments Renewal and Phototransduction Due to a Peripherin Rare Haplotype Variant: Insights from Molecular Modeling. Int. J. Mol. Sci. 2021, 22, 3484. [Google Scholar] [CrossRef]
- Wells, J.; Wroblewski, J.; Keen, J.; Inglehearn, C.; Jubb, C.; Eckstein, A.; Jay, M.; Arden, G.; Bhattacharya, S.; Fitzke, F.; et al. Mutations in the human retinal degeneration slow (RDS) gene can cause either retinitis pigmentosa or macular dystrophy. Nat. Genet. 1993, 3, 213–218. [Google Scholar] [CrossRef]
- Roosing, S.; Thiadens, A.A.; Hoyng, C.B.; Klaver, C.C.; den Hollander, A.I.; Cremers, F.P. Causes and consequences of inherited cone disorders. Prog. Retin. Eye Res. 2014, 42, 1–26. [Google Scholar] [CrossRef]
- Leroy, B.P.; Kailasanathan, A.; De Laey, J.J.; Black, G.C.; Manson, F.D. Intrafamilial phenotypic variability in families with RDS mutations: Exclusion of ROM1 as a genetic modifier for those with retinitis pigmentosa. Br. J. Ophthalmol. 2007, 91, 89–93. [Google Scholar] [CrossRef] [Green Version]
- Daftarian, N.; Mirrahimi, M.; Sabbaghi, H.; Moghadasi, A.; Zal, N.; Dehghan Banadaki, H.; Ahmadieh, H.; Suri, F. PRPH2 mutation as the cause of various clinical manifestations in a family affected with inherited retinal dystrophy. Ophthalmic. Genet. 2019, 40, 436–442. [Google Scholar] [CrossRef] [PubMed]
- Pontikos, N.; Arno, G.; Jurkute, N.; Schiff, E.; Ba-Abbad, R.; Malka, S.; Gimenez, A.; Georgiou, M.; Wright, G.; Armengol, M.; et al. Genetic Basis of Inherited Retinal Disease in a Molecularly Characterized Cohort of More Than 3000 Families from the United Kingdom. Ophthalmology 2020, 127, 1384–1394. [Google Scholar] [CrossRef]
- Conley, S.M.; Stuck, M.W.; Watson, J.N.; Naash, M.I. Rom1 converts Y141C-Prph2-associated pattern dystrophy to retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 509–518. [Google Scholar] [CrossRef]
- Poloschek, C.M.; Bach, M.; Lagreze, W.A.; Glaus, E.; Lemke, J.R.; Berger, W.; Neidhardt, J. ABCA4 and ROM1: Implications for modification of the PRPH2-associated macular dystrophy phenotype. Invest. Ophthalmol. Vis. Sci. 2010, 51, 4253–4265. [Google Scholar] [CrossRef] [Green Version]
- Martin-Merida, I.; Aguilera-Garcia, D.; Fernandez-San Jose, P.; Blanco-Kelly, F.; Zurita, O.; Almoguera, B.; Garcia-Sandoval, B.; Avila-Fernandez, A.; Arteche, A.; Minguez, P.; et al. Toward the Mutational Landscape of Autosomal Dominant Retinitis Pigmentosa: A Comprehensive Analysis of 258 Spanish Families. Invest. Ophthalmol. Vis. Sci. 2018, 59, 2345–2354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birtel, J.; Eisenberger, T.; Gliem, M.; Muller, P.L.; Herrmann, P.; Betz, C.; Zahnleiter, D.; Neuhaus, C.; Lenzner, S.; Holz, F.G.; et al. Clinical and genetic characteristics of 251 consecutive patients with macular and cone/cone-rod dystrophy. Sci. Rep. 2018, 8, 4824. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.S.; Georgiou, M.; Kalitzeos, A.; Moore, A.T.; Michaelides, M. Progressive cone and cone-rod dystrophies: Clinical features, molecular genetics and prospects for therapy. Br. J. Ophthalmol. 2019, 103, 711–720. [Google Scholar] [CrossRef] [Green Version]
- Manes, G.; Guillaumie, T.; Vos, W.L.; Devos, A.; Audo, I.; Zeitz, C.; Marquette, V.; Zanlonghi, X.; Defoort-Dhellemmes, S.; Puech, B.; et al. High prevalence of PRPH2 in autosomal dominant retinitis pigmentosa in france and characterization of biochemical and clinical features. Am. J. Ophthalmol. 2015, 159, 302–314. [Google Scholar] [CrossRef]
- Gao, F.J.; Li, J.K.; Chen, H.; Hu, F.Y.; Zhang, S.H.; Qi, Y.H.; Xu, P.; Wang, D.D.; Wang, L.S.; Chang, Q.; et al. Genetic and Clinical Findings in a Large Cohort of Chinese Patients with Suspected Retinitis Pigmentosa. Ophthalmology 2019, 126, 1549–1556. [Google Scholar] [CrossRef] [Green Version]
- Yoon, C.K.; Kim, N.K.; Joung, J.G.; Shin, J.Y.; Park, J.H.; Eum, H.H.; Lee, H.O.; Park, W.Y.; Yu, H.G. The diagnostic application of targeted re-sequencing in Korean patients with retinitis pigmentosa. BMC Genom. 2015, 16, 515. [Google Scholar] [CrossRef] [Green Version]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and usher syndrome patients by next-generation sequencing. Invest. Ophthalmol. Vis. Sci. 2014, 55, 7369–7375. [Google Scholar] [CrossRef]
- Koyanagi, Y.; Akiyama, M.; Nishiguchi, K.M.; Momozawa, Y.; Kamatani, Y.; Takata, S.; Inai, C.; Iwasaki, Y.; Kumano, M.; Murakami, Y.; et al. Genetic characteristics of retinitis pigmentosa in 1204 Japanese patients. J. Med. Genet. 2019, 56, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Maeda, A.; Yoshida, A.; Kawai, K.; Arai, Y.; Akiba, R.; Inaba, A.; Takagi, S.; Fujiki, R.; Hirami, Y.; Kurimoto, Y.; et al. Development of a molecular diagnostic test for Retinitis Pigmentosa in the Japanese population. Jpn. J. Ophthalmol. 2018, 62, 451–457. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Next-generation sequencing-based comprehensive molecular analysis of 43 Japanese patients with cone and cone-rod dystrophies. Mol. Vis. 2016, 22, 150–160. [Google Scholar] [PubMed]
- Tsunoda, K. Investigations of gene pathogenicity and establishment of a patient data bank for the hereditary chorioretinal dystrophy. Nippon Ganka Gakkai Zasshi 2020, 124, 247–284. [Google Scholar]
- Marmor, M.F.; Fulton, A.B.; Holder, G.E.; Miyake, Y.; Brigell, M.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2008 update). Doc. Ophthalmol. 2009, 118, 69–77. [Google Scholar] [CrossRef] [Green Version]
- McCulloch, D.L.; Marmor, M.F.; Brigell, M.G.; Hamilton, R.; Holder, G.E.; Tzekov, R.; Bach, M. ISCEV Standard for full-field clinical electroretinography (2015 update). Doc. Ophthalmol. 2015, 130, 1–12. [Google Scholar] [CrossRef]
- Constable, P.A.; Bach, M.; Frishman, L.J.; Jeffrey, B.G.; Robson, A.G. ISCEV Standard for clinical electro-oculography (2017 update). Doc. Ophthalmol. 2017, 134, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Fujinami-Yokokawa, Y.; Fujinami, K.; Kuniyoshi, K.; Hayashi, T.; Ueno, S.; Mizota, A.; Shinoda, K.; Arno, G.; Pontikos, N.; Yang, L.; et al. Clinical and Genetic Characteristics of 18 Patients from 13 Japanese Families with CRX-associated retinal disorder: Identification of Genotype-phenotype Association. Sci. Rep. 2020, 10, 9531. [Google Scholar] [CrossRef]
- Fujinami, K.; Kameya, S.; Kikuchi, S.; Ueno, S.; Kondo, M.; Hayashi, T.; Shinoda, K.; Machida, S.; Kuniyoshi, K.; Kawamura, Y.; et al. Novel RP1L1 Variants and Genotype-Photoreceptor Microstructural Phenotype Associations in Cohort of Japanese Patients With Occult Macular Dystrophy. Invest. Ophthalmol. Vis. Sci. 2016, 57, 4837–4846. [Google Scholar] [CrossRef] [Green Version]
- Numa, S.; Oishi, A.; Higasa, K.; Oishi, M.; Miyata, M.; Hasegawa, T.; Ikeda, H.O.; Otsuka, Y.; Matsuda, F.; Tsujikawa, A. EYS is a major gene involved in retinitis pigmentosa in Japan: Genetic landscapes revealed by stepwise genetic screening. Sci. Rep. 2020, 10, 20770. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, K.; Oishi, A.; Yang, L.; Arno, G.; Pontikos, N.; Yoshitake, K.; Fujinami-Yokokawa, Y.; Liu, X.; Hayashi, T.; Katagiri, S.; et al. Clinical and genetic characteristics of 10 Japanese patients with PROM1-associated retinal disorder: A report of the phenotype spectrum and a literature review in the Japanese population. Am. J. Med Genet. Part. C Semin. Med Genet. 2020, 184, 656–674. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.; Feltgen, N.; Junker, B.; Schulze-Bonsel, K.; Bach, M. Resolving the clinical acuity categories "hand motion" and "counting fingers" using the Freiburg Visual Acuity Test (FrACT). Graefes Arch. Clin. Exp. Ophthalmol. 2009, 247, 137–142. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Mizobuchi, K.; Yoshitake, K.; Iwata, T.; Nakano, T. Autosomal dominant retinitis pigmentosa with macular involvement associated with a disease haplotype that included a novel PRPH2 variant (p.Cys250Gly). Ophthalmic. Genet. 2018, 39, 357–365. [Google Scholar] [CrossRef]
- Miyata, M.; Oishi, A.; Oishi, M.; Hasegawa, T.; Ikeda, H.O.; Tsujikawa, A. Long-term efficacy and safety of anti-VEGF therapy in retinitis pigmentosa: A case report. BMC Ophthalmol. 2018, 18, 248. [Google Scholar] [CrossRef]
- Meins, M.; Grüning, G.; Blankenagel, A.; Krastel, H.; Reck, B.; Fuchs, S.; Schwinger, E.; Gal, A. Heterozygous ‘Null Allele’ Mutation in the Human Peripherin/RDS Gene. Hum. Mol. Genet. 1993, 2, 2181–2182. [Google Scholar] [CrossRef]
- Hoyng, C.B.; Heutink, P.; Testers, L.; Pinckers, A.; Deutman, A.F.; Oostra, B.A. Autosomal Dominant Central Areolar Choroidal Dystrophy Caused by a Mutation in Codon 142 in the Peripherin/RDS Gene. Am. J. Ophthalmol. 1996, 121, 623–629. [Google Scholar] [CrossRef] [Green Version]
- Testa, F.; Marini, V.; Rossi, S.; Interlandi, E.; Nesti, A.; Rinaldi, M.; Varano, M.; Garré, C.; Simonelli, F. A Novel Mutation in the RDS Gene in an Italian Family with Pattern Dystrophy. Br. J. Ophthalmol. 2005, 89, 1066–1068. [Google Scholar] [CrossRef] [Green Version]
- Kohl, S.; Giddings, I.; Besch, D.; Apfelstedt-Sylla, E.; Zrenner, E.; Wissinger, B. The Role of the Peripherin/RDS Gene in Retinal Dystrophies. Acta Anat. 1998, 162, 75–84. [Google Scholar] [CrossRef]
- Nakazawa, M.; Kikawa, E.; Chida, Y.; Wada, Y.; Shiono, T.; Tamai, M. Autosomal dominant cone-rod dystrophy associated with mutations in codon 244 (Asn244His) and codon 184 (Tyr184Ser) of the peripherin/RDS gene. Arch. Ophthalmol. 1996, 114, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.J.; Goetz, K.E.; Guan, B.; Ullah, E.; Blain, D.; Zein, W.M.; Tumminia, S.J.; Hufnagel, R.B. Genotype-phenotype associations in a large PRPH2-related retinopathy cohort. Hum. Mutat. 2020, 41, 1528–1539. [Google Scholar] [CrossRef]
- Nakazawa, M.; Naoi, N.; Wada, Y.; Nakazaki, S.; Maruiwa, F.; Sawada, A.; Tamai, M. Autosomal dominant cone-rod dystrophy associated with a Val200Glu mutation of the peripherin/RDS gene. Retina 1996, 16, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.J.; Martinez-Gimeno, M.; Giménez, A.; Lorda, I.; Bueno, J.; García-Sandoval, B.; Ramos, C.; Carballo, M.; Ayuso, C. Two Novel Mutations (Y141H; C214Y) and Previously Published Mutation (R142W) in the RDS-Peripherin Gene in Autosomal Dominant Macular Dystrophies in Spanish Families. Hum. Mutat. 2001, 17, 80. [Google Scholar] [CrossRef]
- Peeters, M.; Khan, M.; Rooijakkers, A.; Mulders, T.; Haer-Wigman, L.; Boon, C.J.F.; Klaver, C.C.W.; van den Born, L.I.; Hoyng, C.B.; Cremers, F.P.M.; et al. PRPH2 mutation update: In silico assessment of 245 reported and 7 novel variants in patients with retinal disease. Hum. Mutat. 2021. [Google Scholar] [CrossRef] [PubMed]
- Boon, C.J.; van Schooneveld, M.J.; den Hollander, A.I.; van Lith-Verhoeven, J.J.C.; Zonneveld-Vrieling, M.N.; Theelen, T.; Cremers, F.P.M.; Hoyng, C.B.; Klevering, B.J. Mutations in the peripherin/RDS gene are an important cause of multifocal pattern dystrophy simulating STGD1/fundus flavimaculatus. Brit. J. Ophthalmol. 2007, 91, 1504–1511. [Google Scholar] [CrossRef] [Green Version]
- Coco-Martin, R.M.; Sanchez-Tocino, H.T.; Desco, C.; Usategui-Martin, R.; Telleria, J.J. PRPH2-Related Retinal Diseases: Broadening the Clinical Spectrum and Describing a New Mutation. Genes 2020, 11, 773. [Google Scholar] [CrossRef] [PubMed]
- Shankar, S.P.; Birch, D.G.; Ruiz, R.S.; Hughbanks-Wheaton, D.K.; Sullivan, L.S.; Bowne, S.J.; Stone, E.M.; Daiger, S.P. Founder Effect of a c.828+3A>T Splice Site Mutation in Peripherin 2 (PRPH2) Causing Autosomal Dominant Retinal Dystrophies. JAMA Ophthalmol. 2015, 133, 511–517. [Google Scholar] [CrossRef]
- Nakazawa, M.; Wada, Y.; Tamai, M. Macular dystrophy associated with monogenic Arg172Trp mutation of the peripherin/RDS gene in a Japanese family. Retina 1995, 15, 518–523. [Google Scholar] [CrossRef]
- Budu; Hayasaka, S.; Matsumoto, M.; Yamada, T.; Zhang, X.Y.; Hayasaka, Y. Peripherin/RDS gene mutation (Pro210Leu) and polymorphisms in Japanese patients with retinal dystrophies. Jpn. J. Ophthalmol. 2001, 45, 355–358. [Google Scholar] [CrossRef]
- Fujiki, K.; Hotta, Y.; Hayakawa, M.; Fujimaki, T.; Takeda, M.; Isashiki, Y.; Ohba, N.; Kanai, A. Analysis of peripherin/RDS gene for Japanese retinal dystrophies. Jpn J. Ophthalmol. 1998, 42, 186–192. [Google Scholar] [CrossRef]
- Yanagihashi, S.; Nakazawa, M.; Kurotaki, J.; Sato, M.; Miyagawa, Y.; Ohguro, H. Autosomal dominant central areolar choroidal dystrophy and a novel Arg195Leu mutation in the peripherin/RDS gene. Arch. Ophthalmol. 2003, 121, 1458–1461. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, M.; Kikawa, E.; Kamio, K.; Chida, Y.; Shiono, T.; Tamai, M. Ocular findings in patients with autosomal dominant retinitis pigmentosa and transversion mutation in codon 244 (Asn244Lys) of the peripherin/RDS gene. Arch. Ophthalmol. 1994, 112, 1567–1573. [Google Scholar] [CrossRef]
- Nakazawa, M.; Kikawa, E.; Chida, Y.; Tamai, M. Asn244His mutation of the peripherin/RDS gene causing autosomal dominant cone-rod degeneration. Hum. Mol. Genet. 1994, 3, 1195–1196. [Google Scholar] [CrossRef] [PubMed]
- Kikawa, E.; Nakazawa, M.; Chida, Y.; Shiono, T.; Tamai, M. A novel mutation (Asn244Lys) in the peripherin/RDS gene causing autosomal dominant retinitis pigmentosa associated with bull’s-eye maculopathy detected by nonradioisotopic SSCP. Genomics 1994, 20, 137–139. [Google Scholar] [CrossRef] [PubMed]
- Saga, M.; Mashima, Y.; Akeo, K.; Oguchi, Y.; Kudoh, J.; Shimizu, N. A novel Cys-214-Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Hum. Genet. 1993, 92, 519–521. [Google Scholar] [CrossRef]
- Gocho, K.; Akeo, K.; Itoh, N.; Kameya, S.; Hayashi, T.; Katagiri, S.; Gekka, T.; Ohkuma, Y.; Tsuneoka, H.; Takahashi, H. High-Resolution Adaptive Optics Retinal Image Analysis at Early Stage Central Areolar Choroidal Dystrophy With PRPH2 Mutation. Ophthalmic Surg. Lasers Imaging Retin. 2016, 47, 1115–1126. [Google Scholar] [CrossRef]
- Marano, F.; Deutman, A.F.; Leys, A.; Aandekerk, A.L. Hereditary retinal dystrophies and choroidal neovascularization. Graefes Arch. Clin. Exp. Ophthalmol. 2000, 238, 760–764. [Google Scholar] [CrossRef]
- Conley, S.M.; Naash, M.I. Gene therapy for PRPH2-associated ocular disease: Challenges and prospects. Cold Spring Harb Perspect. Med. 2014, 4, a017376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falfoul, Y.; Matri, K.E.; Habibi, I.; Halouani, S.; Chebil, A.; Schorderet, D.; El Matri, L. OCT-angiography assessing quiescent and active choroidal neovascularization in retinitis pigmentosa associated with PRPH2 pathogenic variant. Eur J. Ophthalmol. 2021. [Google Scholar] [CrossRef] [PubMed]
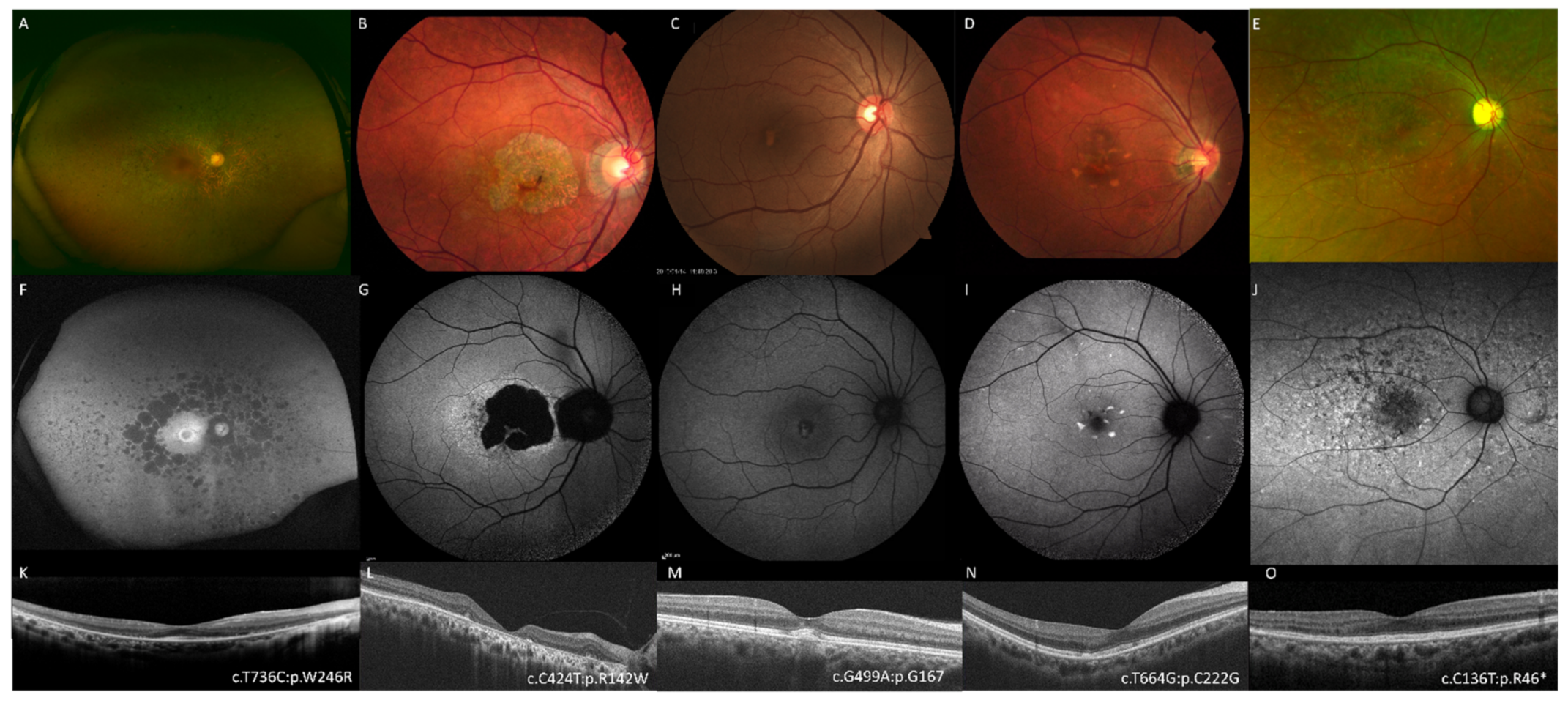




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Family ID | Inheritance Trait | Sex | Age | Onset Age | Visual Acuity * Right/Left | Macular Atrophy | Peripheral Atrophy | Best Disease-Like Deposit | Flecks | Phenotype SubGroup | Variants |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NISO 1034-001 | 1 | AD | F | 57 | 10 | 0.05/0.00 | − | + | − | − | RP | c.136C>T (p.R46 *) |
| UOEH 188-2 | 2 | AD | F | 42 | Unknown | 0.00/−0.08 | + | − | − | + | MD | c.136C>T (p.R46 *) |
| UOEH 188-1 | 2 | AD | F | 67 | Unknown | 0.00/0.00 | + | − | − | + | MD | c.136C>T (p.R46 *) |
| KYT 6074 | 3 | AD | F | 68 | 43 | 0.00/0.15 | + | + | − | − | RP | c.410G>A (p.G137D) |
| KYT 6322 | 4 | AD | M | 53 | 46 | 0.15/0.10 | − | + | − | − | RP | c.410G>A (p.G137D) |
| MYZ 098-001 | 5 | AD | F | 42 | No symptom | 0.05/0.05 | + | − | − | − | CRD | c.424C>T (p.R142W) |
| MYZ 098-002 | 5 | AD | M | 69 | 58 | 2.30/2.30 | + | + | − | − | RD | c.424C>T (p.R142W) |
| KYT 6703 | 6 | AD | M | 73 | Unknown | 1.10/1.10 | + | − | − | − | MD | c.424C>T (p.R142W) |
| KYT 4296 | 6 | AD | M | 78 | 70 | 1.30/0.22 | + | − | − | − | MD | c.424C>T (p.R142W) |
| KND 129-75 | 7 | Sporadic | F | 56 | Unknown | 0.15/0.15 | + | − | − | − | MD | c.424C>T (p.R142W) |
| NISO 1014-001 | 8 | Sporadic | M | 64 | 58 | 0.15/0.00 | + | − | − | − | MD | c.424C>T (p.R142W) |
| KYT 6274 | 9 | AD | F | 61 | 41 | 0.00/0.70 | − | + | − | − | RP | c.499G>A (p.G167S) |
| KYT 6144 | 9 | AD | F | 88 | Unknown | 0.00/0.05 | + | − | − | + | MD | c.499G>A (p.G167S) |
| JKI 167-1314 | 10 | Sporadic | F | 50 | Unknown | −0.08/0.00 | − | − | + | − | MD | c.499G>A (p.G167S) |
| MYZ 040-001 | 11 | Sporadic | F | 75 | 60 | 0.52/0.22 | − | + | − | − | RP | c.508G>A, (p.G170S) |
| NGY 0255 | 12 | n.a. | M | 43 | Unknown | 0.52/0.40 | + | − | − | − | CRD | c.514C>T, (p.R172W) |
| NISO 1010-001 | 13 | Sporadic | M | 52 | 45 | 0.22/0.05 | + | − | − | + | MD | c.514C>T, (p.R172W) |
| NGY 0196 | 14 | Sporadic | M | 62 | 60 | 0.40/0.22 | + | − | − | − | MD | c.514C>T, (p.R172W) |
| JKI 275-1927 | 15 | Sporadic | M | 57 | 56 | 0.30/0.52 | + | − | − | − | MD | c.514C>T, (p.R172W) |
| KB 001-01 | 16 | AD | M | 66 | 46 | 0.22/0.70 | + | ; | − | − | RP | c.551A>C (p.Y184S) |
| KYT 6399 | 17 | AD | F | 73 | 25 | 1.70/1.70 | + | − | − | − | MD | c.589A>G (p.K197E) |
| MYZ 093-001 | 18 | AD | M | 61 | 23 | 2.30/2.30 | + | + | − | − | RP | c.599T>A (p.V200E) |
| MYZ 093-002 | 18 | AD | M | 64 | 45 | 1.52/2.00 | + | + | − | − | RP | c.599T>A (p.V200E) |
| MYZ 093-004 | 18 | AD | M | 33 | 20 | 0.00/0.00 | + | − | − | − | RP | c.599T>A (p.V200E) |
| MYZ 093-005 | 18 | AD | M | 31 | 28 | −0.08/−0.08 | + | − | − | − | CRD | c.599T>A (p.V200E) |
| MYZ 101-001 | 19 | AD | F | 32 | 29 | 0.30/0.30 | + | − | − | − | CRD | c.599T>A (p.V200E) |
| MYZ 101-003 | 19 | AD | F | 52 | 5 | 1.30/1.22 | + | − | − | − | CRD | c.599T>A (p.V200E) |
| MYZ 066-001 | 20 | AD | F | 40 | 32 | 0.40/0.52 | + | + | − | − | CRD | c.599T>A (p.V200E) |
| NGY 0148 | 21 | AD | M | 49 | Unknown | 0.00/0.00 | + | − | − | + | MD | c.641G>A (p.C214Y) |
| NISO 112-112 | 22 | Sporadic | M | 49 | 14 | −0.08/0.00 | + | + | − | − | MD | c.664T>G (p.C222G) |
| KND 109-38 | 23 | AD | F | 28 | 8 | 0.00/0.00 | − | + | − | − | RP | c.670_681del (p.Q224_I227del) |
| KND 109-51 | 23 | AD | M | 30 | 14 | −0.08/−0.08 | − | + | − | − | RP | c.670_681del (p.Q224_I227del) |
| KND 109-52 | 23 | AD | M | 67 | 27 | 2.00/1.52 | + | + | − | − | RP | c.670_681del (p.Q224_I227del) |
| KYT 6553 | 24 | AD | F | 58 | 15 | 0.05/0.05 | − | + | − | − | RP | c.736T>C (p.W246R) |
| JKI 060-0665 | 25 | AD | M | 48 | 45 | −0.08/−0.18 | − | − | + | − | RP | c.748T>G (p.C250G) |
| KYT 6237 | 26 | n.a. | F | 49 | Unknown | −0.18/0.05 | − | − | + | − | MD | c.748T>G (p.C250G) |
| KYT 6102 | 27 | Sporadic | F | 53 | 37 | 0.52/-0.08 | + | − | − | + | MD | c.748T>G (p.C250G) |
| JKI 077-0735 | 28 | Sporadic | M | 58 | 56 | −0.08/0.00 | + | − | + | + | MD | c.809_810del (p.L270Pfs *30) |
| NISO 4002-001 | 29 | Sporadic | F | 38 | 6 | 1.70/1.52 | − | + | − | − | RP | c.828G>T (p.E276D) |
| NISO 3001-001 | 30 | AD/AR | F | 59 | 50 | 0.52/0.52 | + | + | − | − | RP | c.828_828+5 delGGTAGGinsC |
| Varaiant ID | Family ID | Nucleotide Change, Amino Acid Change | Position (hg19) | Coding Impact | dbSNP ID | HGVD | iJGVD 3.5k | 1000 Genome | GnomAD Allele Frequency | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Allele Frequency | Coverage in GnomAD Exomes Samples | ||||||||||||||||
| East Asian | South Asian | African | European (Non-Finnish) | Total | Mean Coverage | Median Coverage | % of Samples Over 20× Coverage | Reference | |||||||||
| 1 | 1,2 | c.136C>T, (p.Arg46Ter) | Chr6:42689937 | Nonsense | rs61755771 | 0.0000% | 0.0000% | NA | NA | NA | 0.0062% | 0.0009% | 0.0035% | 95.9 | 100 | 99.88% | Meins (1993) Hum Mol Genet. [38] |
| 2 | 3, 4 | c.410G>A, (p.Gly137Asp) | Chr6:42689663 | Missense | rs527236097 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 87.2 | 100 | 99.90% | Novel (listed on ClinVar, SCV001240217.1) |
| 3 | 5, 6, 7, 8 | c.424C>T, (p.Arg142Trp) | Chr6:42689649 | Missense | rs61755783 | 0.0000% | 0.0000% | NA | NA | NA | NA | 0.0009% | 0.0016% | 89.3 | 100 | 99.30% | Hoyng (1996) Am J Ophthalmol [39] |
| 4 | 9, 10 | c.499G>A, (p.Gly167Ser) | Chr6:42689574 | Missense | rs527236098 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 98.9 | 100 | 100.00% | Testa (2005) Br J Ophthalmol [40] |
| 5 | 11 | c.508G>A, (p.Gly170Ser) | Chr6:42689565 | Missense | rs61755791 | 0.0000% | 0.0000% | NA | 0.0163% | 0.0033% | NA | NA | 0.0016% | 99.2 | 100 | 99.99% | Kohl (1998) Acta Anat (Basel) [41] |
| 6 | 12, 13, 14, 15 | c.514C>T, (p.Arg172Trp) | Chr6:42689559 | Missense | rs61755792 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 99.2 | 100 | 99.97% | Wells (1993) Nat Genet [9] |
| 7 | 16 | c.551A>C, (p.Tyr184Ser) | Chr6:42689522 | Missense | rs62645926 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 99.1 | 100 | 99.89% | Nakazawa (1996) Arch Ophthalmol [42] |
| 8 | 17 | c.589A>G, (p.Lys197Glu) | Chr6:42672342 | Missense | rs62645931 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 89.0 | 100 | 99.88% | Reeves (2020) Hum Mutat [43] |
| 9 | 18, 19, 20 | c.599T>A, (p.Val200Glu) | Chr6:42672332 | Missense | rs62645932 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 89.4 | 100 | 99.89% | Nakazawa (1996) Retina [44] |
| 10 | 21 | c.641G>A, (p.Cys214Tyr) | Chr6:42672290 | Missense | rs61755804 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 87.1 | 100 | 99.96% | Trujillo (2000) Hum Mutat [45] |
| 11 | 22 | c.664T>G, (p.Cys222Gly) | Chr6:42672267 | Missense | NA | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 82.0 | 100 | 99.87% | Novel |
| 12 | 23 | c.670_681del, (p.Gln224_Ile227del) | Chr6:42672250 | In frame | NA | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 78.8 81.3 | 100 100 | 99.59% 99.85% | Novel |
| 13 | 24 | c.736T>C, (p.Trp246Arg) | Chr6:42672195 | Missense | rs61755817 | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 69.2 | 100 | 89.02% | Novel |
| 14 | 25, 26, 27 | c.748T>G, (p.Cys250Gly) | Chr6:42672183 | Missense | NA | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 67.6 | 100 | 82.58% | Katagiri (2018) Ophthalmic Genet [36] |
| 16 | 28 | c.809_810delT, (p.Leu270ProfsTer30) | Chr6:42672121 | Frameshift | NA | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 64.4 64.4 | 100 100 | 68.64% 68.72% | Peeters (2021) Hum Mutat [46] |
| 17 | 29 | c.828G>T, (p.Glu276Asp) | Chr6:42672103 | Missense (splice site alteration) | NA | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 63.6 | 100 | 65.95% | Novel |
| 18 | 30 | c.828_828+5delGGTAGGinsC | Chr6:42672098 | Splice site alteration | NA | 0.0000% | 0.0000% | NA | NA | NA | NA | NA | NA | 63.3 63.6 | 100 100 | 65.21% 65.95% | Novel |
| Variant ID | Family ID | Nucleotide Change, Amino Acid Change | General Prediction | Functional Prediction | Conservation | ACMG Classification | ||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MutationTaster | FATHMM | SIFT | PROVEAN | Polyphen2 | PhyloP30way | PhastCons30way | Verdict | Classification Categories | ||||||||||||||||||||
| Prediction | Accuruacy | Converted Rankscore | Prediction | Score | Converted Rankscore | Prediction | Score | Converted Rankscore | Prediction | Score | Converted Rankscore | Prediction | Score | Mammalian | Mammalian Rankscore | Mammalian | Mammalian Rankscore | |||||||||||
| 1 | 1, 2 | c.136C>T, p.(Arg46*) | Disease causing automatic | 1 | 0.81 | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 1.1759 | 0.7892 | 0.986 | 0.5005 | Pathogenic | PVS1 (strong) | PM2 | PP1 | PP3 | PP5 | ||
| 2 | 3, 4 | c.410G>A, (p.Gly137Asp) | Disease causing | 0.9997 | 0.49 | Damaging | −2.2 | 0.8689 | Damaging | 0.003 | 0.6824 | Damaging | −2.83 | 0.5971 | Probably Damaging | 0.963 | 1.026 | 0.4595 | 0.9629 | 0.4369 | Likely Pathogenic | PM1 | PM2 | PP1 | PP3 | PP5 | ||
| 3 | 5, 6, 7, 8 | c.424C>T, p.(Arg142Trp) | Disease causing automatic | 0.9304 | 0.37 | Tolerated | −1.28 | 0.7938 | Damaging | 0.000 | 0.9125 | Damaging | −4.08 | 0.7474 | Probably Damaging | 0.998 | 0.2119 | 0.2406 | 0.9969 | 0.6203 | Pathogenic | PM1 | PM2 | PM5 | PP1 | PP3 | PP5 | |
| 4 | 9, 10 | c.499G>A, p.(Gly167Ser) | Disease causing | 1 | 0.81 | Damaging | −5.54 | 0.9918 | Damaging | 0.000 | 0.9125 | Damaging | −5.29 | 0.8432 | Probably Damaging | 1.000 | 1.026 | 0.4595 | 0.762 | 0.3244 | Pathogenic | PS1 | PM1 | PM2 | PM5 | PP1 | PP3 | PP5 |
| 5 | 11 | c.508G>A, p.(Gly170Ser) | Disease causing | 0.9998 | 0.49 | Damaging | −2.24 | 0.8719 | Tolerated | 0.100 | 0.3052 | Neutral | −1.13 | 0.2911 | Possibly Damaging | 0.822 | 1.026 | 0.4595 | 0.949 | 0.4173 | Likely Pathogenic | PM1 | PM2 | PP3 | PP5 | |||
| 6 | 12, 13, 14, 15 | c.514C>T, p.(Arg172Trp) | Disease causing | 0.962% | 0.38 | Tolerated | −1.36 | 0.8012 | Damaging | 0.000 | 0.9125 | Damaging | −5.33 | 0.8460 | Probably Damaging | 0.999 | 1.1759 | 0.7892 | 0.9919 | 0.5410 | Pathogenic | PM1 | PM2 | PM5 | PP3 | PP5 | \ | |
| 7 | 16 | c.551A>C, p.(Tyr184Ser) | Disease causing | 1 | 0.81 | Tolerated | −1.13 | 0.7772 | Damaging | 0.000 | 0.9125 | Damaging | −6.16 | 0.9021 | Probably Damaging | 1.000 | 1.138 | 0.6469 | 1 | 0.8628 | Likely Pathogenic | PM1 | PM2 | PP2 | PP5 | |||
| 8 | 17 | c.589A>G, (p.Lys197Glu) | Disease causing | 0.9953 | 0.43 | Tolerated | 4.1 | 0.0294 | Damaging | 0.035 | 0.4371 | NA | NA | NA | Probably Damaging | 0.759 | 1.138 | 0.6469 | 1 | 0.8628 | Likely Pathogenic | PM1 | PM2 | PP3 | PP5 | |||
| 9 | 18, 19, 20 | c.599T>A, p.(Val200Glu) | Disease causing | 1 | 0.81 | Tolerated | 3.88 | 0.0357 | Damaging | 0.000 | 0.9125 | Damaging | −3.85 | 0.7235 | Possibly Damaging | 0.941 | 1.312 | 0.9471 | 1 | 0.8628 | Likely Pathogenic | PM1 | PM2 | PP1 | PP3 | PP5 | ||
| 10 | 21 | c.641G>A, p.(Cys214Tyr) | Disease causing | 1 | 0.81 | Damaging | −3.25 | 0.9353 | Damaging | 0.000 | 0.9125 | Damaging | −10.22 | 0.9893 | Probably Damaging | 1.000 | 1.026 | 0.4595 | 0.999 | 0.7043 | Pathogenic | PM1 | PM2 | PM5 | PP3 | PP5 | ||
| 11 | 22 | c.664T>G, p.(Cys222Gly) | Disease causing | 1 | 0.81 | Damaging | −2.08 | 0.8601 | Damaging | 0.000 | 0.9125 | Damaging | −11.10 | 0.9934 | Probably Damaging | 0.995 | 1.312 | 0.9471 | 0.9959 | 0.5952 | Likely Pathogenic | PM1 | PM2 | PM5 | PP3 | |||
| 12 | 23 | c.670_681del, p.(Gln224_Ile227del) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Likely Pathogenic | PM1 | PM2 | PM4 | PP1 | PP3 | ||
| 13 | 24 | c.736T>C, (p.Trp246Arg) | Disease causing | 1 | 0.81 | Tolerated | −1.26 | 0.7918 | Damaging | 0.003 | 0.6824 | Damaging | −7.52 | 0.951 | Probably Damaging | 0.996 | 1.312 | 0.9471 | 0.995 | 0.5772 | Likely Pathogenic | PM1 | PM2 | PM5 | PP3 | |||
| 14 | 25, 26, 27 | c.748T>G, p.(Cys250Gly) | Disease causing | 1 | 0.81 | Damaging | −7.44 | 0.9988 | Damaging | 0.000 | 0.9125 | Damaging | −10.84 | 0.9922 | Probably Damaging | 1.000 | 1.312 | 0.9471 | 0.995 | 0.5772 | Likely Pathogenic | PM1 | PM2 | PM5 | PP3 | PP5 | ||
| 16 | 28 | c.809_810delTC, p.(Leu270Profs*30) | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | 1.307 | 0.8849 | 0.039 | 0.1619 | Likely Pathogenic | PVS1 (moderate) | PM2 | PP3 | PP5 | |||
| 17 | 29 | c.828G>T, p.(Glu276Asp) | Disease causing | 0.9999 | 0.53 | Tolerated | −1.3 | 0.7957 | Damaging | 0.023 | 0.4819 | Neutral | −2.17 | 0.4902 | Possibly Damaging | 0.680 | 1.0219 | 0.3987 | 0.8949 | 0.3737 | Likely Pathogenic | PVS1 (strong) | PM2 | |||||
| 18 | 30 | c.828_828+5delGGTAGGinsC | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | NA | Likely Pathogenic | PVS1 (strong) | PM2 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oishi, A.; Fujinami, K.; Mawatari, G.; Naoi, N.; Ikeda, Y.; Ueno, S.; Kuniyoshi, K.; Hayashi, T.; Kondo, H.; Mizota, A.; et al. Genetic and Phenotypic Landscape of PRPH2-Associated Retinal Dystrophy in Japan. Genes 2021, 12, 1817. https://doi.org/10.3390/genes12111817
Oishi A, Fujinami K, Mawatari G, Naoi N, Ikeda Y, Ueno S, Kuniyoshi K, Hayashi T, Kondo H, Mizota A, et al. Genetic and Phenotypic Landscape of PRPH2-Associated Retinal Dystrophy in Japan. Genes. 2021; 12(11):1817. https://doi.org/10.3390/genes12111817
Chicago/Turabian StyleOishi, Akio, Kaoru Fujinami, Go Mawatari, Nobuhisa Naoi, Yasuhiro Ikeda, Shinji Ueno, Kazuki Kuniyoshi, Takaaki Hayashi, Hiroyuki Kondo, Atsushi Mizota, and et al. 2021. "Genetic and Phenotypic Landscape of PRPH2-Associated Retinal Dystrophy in Japan" Genes 12, no. 11: 1817. https://doi.org/10.3390/genes12111817