
Identification, Analysis, and Confirmation of Seed Storability-Related Loci in Dongxiang Wild Rice (Oryza rufipogon Griff.)

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Seed Storability Evaluation and Extreme Bulk Sample Construction
2.3. Sequence Libraries Construction and BSA-Seq
2.4. Molecular Marker Background Analysis
3. Results
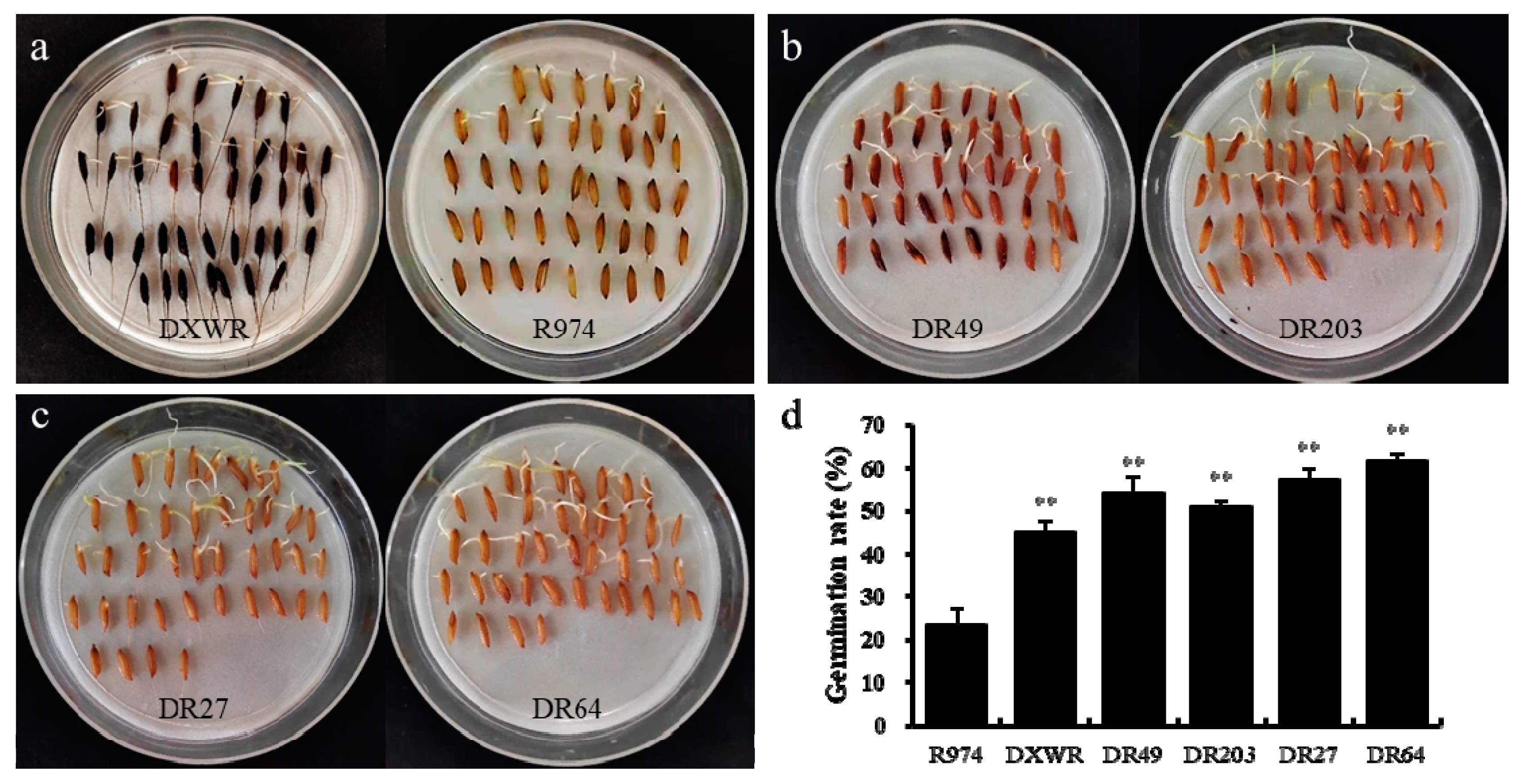
3.1. Identification of BILs with Extreme Phenotypes for Seed Storability
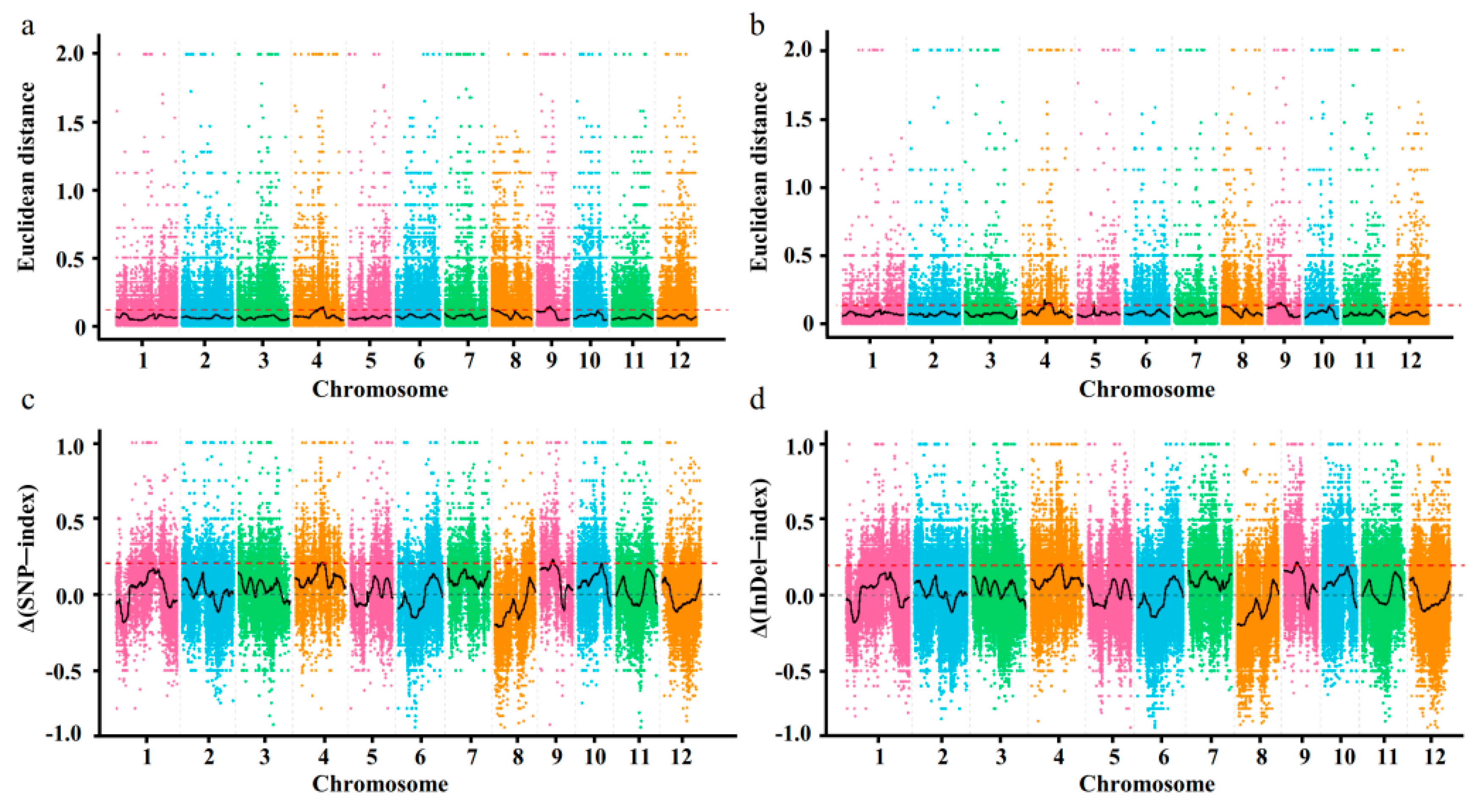
3.2. BSA-Seq, Data Analysis, and Quality Assessment
3.3. Identification of Candidate Genome Loci for Seed Storability
3.4. Bioinformatics Analysis of Annotated Genes in the Identified Regions
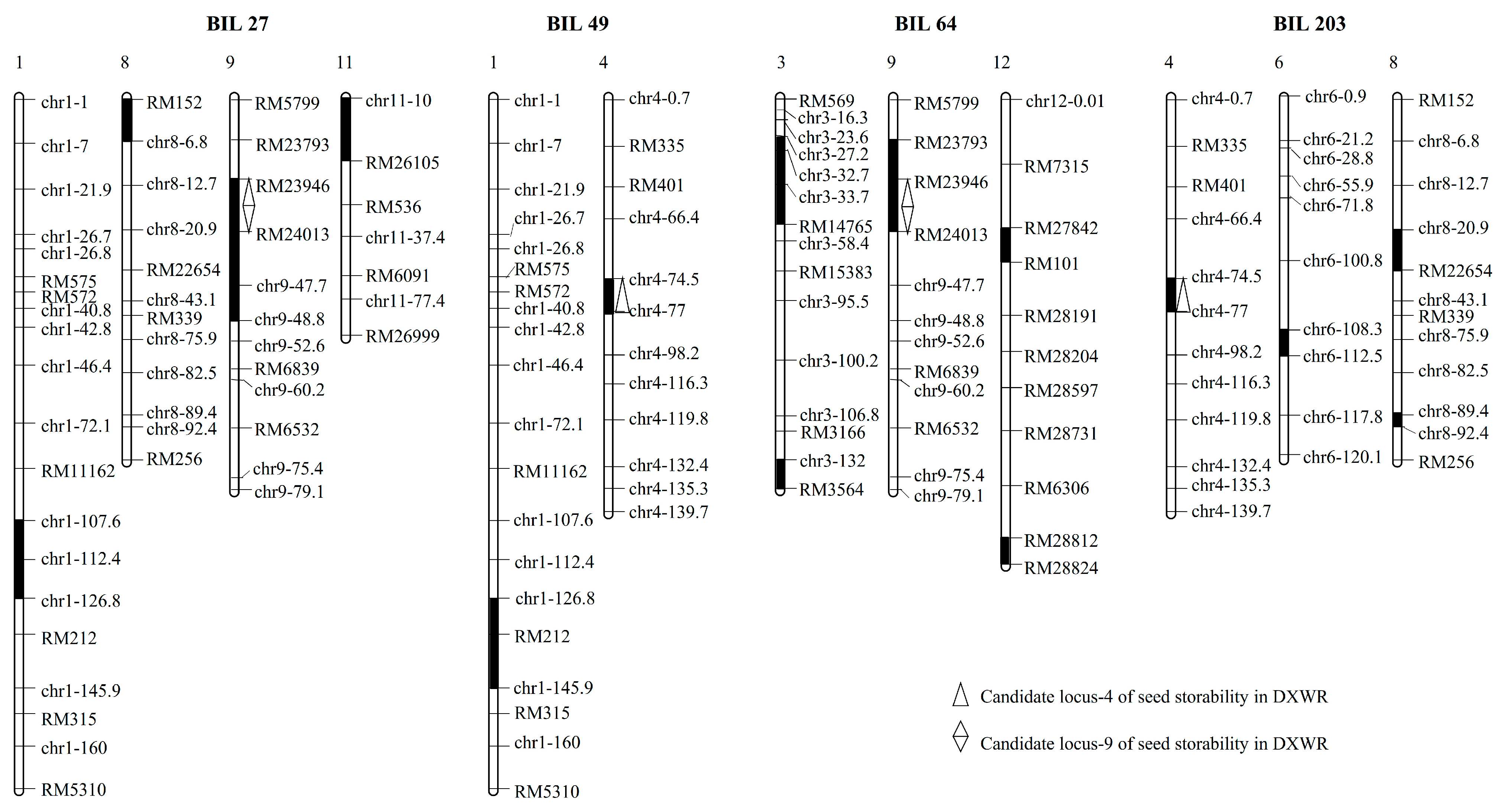
3.5. Confirmation of the Candidate Loci for Seed Storability
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yan, W.G.; Li, Y.; Agrama, H.A.; Luo, D.G.; Gao, F.Y.; Lu, X.J.; Ren, G.J. Association mapping of stigma and spikelet characteristics in rice (Oryza sativa L.). Mol. Breed. 2009, 24, 277–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.L.; Zhang, Y.; Sun, J.; Meng, J.S.; Tao, J. Deterioration of orthodox seeds during ageing: Influencing factors, physiological alterations and the role of reactive oxygen species. Plant Physiol. Biochem. 2020, 158, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.; Huang, W.; Gao, J.; Fu, H.; Liu, J. Comparative metabolomic analysis of seed metabolites associated with seed storability in rice (Oryza sativa L.) during natural aging. Plant Physiol. Biochem. 2018, 127, 590–598. [Google Scholar] [CrossRef]
- Zhang, H.; Duan, Z.; Li, Y.; Zhao, G.; Zhu, S.; Fu, W.; Peng, T.; Zhao, Q.; Svanberg, S.; Hu, J. Vis/NIR reflectance spectroscopy for hybrid rice variety identification and chlorophyll content evaluation for different nitrogen fertilizer levels. Roy. Soc. Open Sci. 2019, 6, 191132. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Zhou, L.; Wei, J.; Tang, Q.; Zou, Y.; Tang, J.; Xu, H. Cover crops and chicken grazing in a winter fallow field improve soil carbon and nitrogen contents and decrease methane emissions. Sci. Rep. 2020, 10, 12607. [Google Scholar] [CrossRef]
- Zhang, P.; Wu, H.M.; Zhang, F.; Cao, P.H.; Cai, M.Y.; Song, W.H.; Liu, S.J.; Jiang, L. Improving the storage tolerance of rice seeds by down-regulating OsLOX by RNAi. J. Nanjing Agric. Univ. 2019, 42, 996–1005, (Chinese with English abstract). [Google Scholar]
- Zhang, X.; Hina, A.; Song, S.; Kong, J.; Bhat, J.A.; Zhao, T. Whole-genome mapping identified novel “QTL hotspots regions” for seed storability in soybean (Glycine max L.). BMC Genom. 2019, 20, 499. [Google Scholar] [CrossRef] [Green Version]
- Harmeet, K.; Petla, B.P.; Kamble, N.U.; Ajeet, S.; Rao, V.; Prafull, S.; Shraboni, G.; Manoj, M. Differentially expressed seed aging responsive heat shock protein OsHSP18.2 implicates in seed vigor; longevity and improves germination and seedling establishment under abiotic stress. Front. Plant Sci. 2015, 6, 713. [Google Scholar]
- He, Y.; Cheng, J.; He, Y.; Yang, B.; Cheng, Y.; Yang, C.; Zhang, H.; Wang, Z. Influence of isopropylmalate synthase OsIPMS1 on seed vigour associated with amino acid and energy metabolism in rice. Plant Biotechnol. J. 2019, 17, 322–337. [Google Scholar] [CrossRef] [Green Version]
- Lei, M.; Zhu, F.; Li, Z.; Zhang, J.; Li, X.; Dong, J.; Wang, T. Talen-based mutagenesis of lipoxygenase Lox3 enhances the storage tolerance of rice (Oryza sativa) seeds. PLoS ONE 2015, 10, e0143877. [Google Scholar]
- Wu, F.; Luo, X.; Wang, L.; Wei, Y.; Li, J.; Xie, H.; Zhang, J.; Xie, G. Genome-wide association study reveals the QTLs for seed storability in world rice core collections. Plants 2021, 10, 812. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, K.; Wu, J.; Guo, N.; Liang, J.; Wang, X.; Cheng, F. QTL-Seq and sequence assembly rapidly mapped the gene BrMYBL2.1 for the purple trait in Brassica rapa. Sci. Rep. 2020, 10, 2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ficiciyan, A.; Loos, J.; Sievers-Glotzbach, S.; Tscharntke, T. More than yield: Ecosystem services of traditional versus modern crop varieties revisited. Sustainability 2018, 10, 2834. [Google Scholar] [CrossRef] [Green Version]
- Liang, T.; Chi, W.; Huang, L.; Qu, M.; Zhang, S.; Chen, Z.Q.; Chen, Z.J.; Tian, D.; Gui, Y.; Chen, X.; et al. Bulked segregant analysis coupled with whole-genome sequencing (BSA-Seq) Mapping identifies a Novel pi21 haplotype conferring basal resistance to rice blast disease. Int. J. Mol. Sci. 2020, 21, 2162. [Google Scholar] [CrossRef] [Green Version]
- Kitony, J.K.; Sunohara, H.; Tasaki, M.; Mori, J.I.; Shimazu, A.; Reyes, V.P.; Yasui, H.; Yamagata, Y.; Yoshimura, A.; Yamasaki, M.; et al. Development of an Aus-derived nested association mapping (Aus-NAM) population in rice. Plants 2021, 10, 1255. [Google Scholar] [CrossRef] [PubMed]
- Reyes, V.P.; Angeles-Shim, R.B.; Mendioro, M.S.; Manuel, M.C.C.; Lapis, R.S.; Shim, J.; Sunohara, H.; Nishiuchi, S.; Kikuta, M.; Makihara, D.; et al. Marker-assisted introgression and stacking of major QTLs controlling grain number (Gn1a) and number of primary branching (WFP) to NERICA cultivars. Plants 2021, 10, 844. [Google Scholar] [CrossRef]
- Wen, J.; Jiang, F.; Weng, Y.; Sun, M.; Shi, X.; Zhou, Y.; Yu, L.; Wu, Z. Identification of heat-tolerance QTLs and high-temperature stress-responsive genes through conventional QTL mapping; QTL-seq and RNA-seq in tomato. BMC Plant Biol. 2019, 19, 398. [Google Scholar] [CrossRef]
- Guo, Z.; Cai, L.; Chen, Z.; Wang, R.; Zhang, L.; Guan, S.; Zhang, S.; Ma, W.; Liu, C.; Pan, G. Identification of candidate genes controlling chilling tolerance of rice in the cold region at the booting stage by BSA-Seq and RNA-Seq. R. Soc. Open Sci. 2020, 7, 201081. [Google Scholar] [CrossRef]
- Lu, H.; Lin, T.; Klein, J.; Wang, S.; Qi, J.; Zhou, Q. QTL-seq identifies an early flowering QTL located near Flowering Locus T in cucumber. Theor. Appl. Genet. 2014, 127, 1491. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Ge, Q.; Zhao, P.; Chen, W.; Sang, X.; Zhao, Y.; Chen, Q.; Wang, H. Rapid mining of candidate genes for verticillium wilt resistance in cotton based on BSA-Seq analysis. Front. Plant Sci. 2021, 12, 703011. [Google Scholar] [CrossRef]
- Zhang, F.; Zhou, Y.; Zhang, M.; Luo, X.; Xie, J. Effects of drought stress on global gene expression profile in leaf and root samples of Dongxiang wild rice (Oryza rufipogon). Biosci. Rep. 2017, 37, BSR20160509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.Y.; Wu, Y.J.; Zhang, C.H.; Jiang, J.P. Preliminary study on storage characteristics of rice seed resources in different evolution stages. Hybrid Rice 2010, 25, 70–72, (Chinese with English abstract). [Google Scholar]
- Ren, R.J.; Wang, P.; Wang, L.N.; Su, J.P.; Sun, L.J.; Sun, Y.; Chen, D.F.; Chen, X.W. Os4BGlu14, a monolignol β-Glucosidase, negatively affects seed longevity by influencing primary metabolism in rice. Plant Mol. Biol. 2020, 104, 513–527. [Google Scholar] [CrossRef]
- Takagi, H.; Tamiru, M.; Abe, A.; Yoshida, K.; Uemura, A.; Yaegashi, H. MutMap accelerates breeding of a salt-tolerant rice cultivar. Nat. Biotechnol. 2015, 33, 445–449. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows Wheeler Transform. Bioinformatics 2009, 25, 160–164. [Google Scholar] [CrossRef] [Green Version]
- Michelmore, R.W.; Paran, I.; Kesseli, R. Identification of markers linked to disease-resistance genes by bulked segregant analysis: A rapid method to detect markers in specific genomic regions by using segregating populations. Proc. Natl. Acad. Sci. USA 1991, 88, 9828–9832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A. The genome analysis toolkit: A mapReduce framework for analyzing next generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Cingolani, P.; Platts, A.; Wangle, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Reumers, J.; De Rijk, P.; Zhao, H.; Liekens, A.; Smeets, D.; Cleary, J.; Van Loo, P.; Van Den Bossche, M.; Catthoor, K.; Sabbe, B.; et al. Optimized filtering reduces the error rate in detecting genomic variants by short-read sequencing. Nat. Biotechnol. 2012, 30, 61–68. [Google Scholar] [CrossRef]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Gorsi, B.; Yost, H.J. MMAPPR: Mutation mapping analysis pipeline for pooled RNA-seq. Genome Res. 2013, 23, 687–697. [Google Scholar] [CrossRef] [Green Version]
- Dokmanic, I.; Parhizkar, R.; Ranieri, J.; Vetterli, M. Euclidean distance matrices: A short walk through theory, algorithms and applications. IEEE Signal. Proc. Mag. 2015, 32, 12–30. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhanag, Z.; Webb, M. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; He, F.C. Integrated nr database in protein annotation system and its localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar]
- Yip, Y.L.; Famiglietti, M.; Gos, A.; Duek, P.D.; David, F.P.A.; Gateau, A.; Bairoch, A. Annotating single amino acid polymorphisms in the UniProt/Swiss-Prot knowledgebase. Hum. Mutat. 2008, 29, 361–366. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, 277–280. [Google Scholar]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, P.; Wang, C.; Huang, K.; Luo, Y.; Xu, W. A novel pretreatment-free duplex chamber digital PCR detection system for the absolute quantitation of gmo samples. Int. J. Mol. Sci. 2016, 17, 402. [Google Scholar] [CrossRef]
- Lin, Q.Y.; Wang, W.Y.; Ren, Y.K.; Jiang, Y.; Sun, A.; Qian, Y.; Zhang, Y.; He, N.; Hang, N.T.; Liu, Z.; et al. Genetic dissection of seed storability using two different populations with a same parent rice cultivar N22. Breed. Sci. 2015, 65, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Fan, K.; Xia, L.; Ding, X.; Tian, L.; Sun, W.; He, H.; Yu, S. Genetic dissection of seed storability and validation of candidate gene associated with antioxidant capability in rice (Oryza sativa L.). Int. J. Mol. Sci. 2019, 20, 4442. [Google Scholar] [CrossRef] [Green Version]
- Quilloy, F.A.; Labaco, B.; Casal, C.; Dixit, S. Crop establishment in direct-seeded rice: Traits, physiology, and genetics. In Rice Improvement; Ali, J., Wani, S.H., Eds.; Springer: Cham, Switzerland, 2021; pp. 171–202. [Google Scholar]
- Hang, N.T.; Lin, Q.Y.; Liu, L.L.; Liu, X. Mapping QTLs related to rice seed storability under natural and artificial aging storage conditions. Euphytica 2015, 203, 673–681. [Google Scholar] [CrossRef]
- Li, C.S.; Shao, G.S.; Wang, L.; Wang, Z.F.; Zhang, H.S. QTL identification and fine mapping for seed storability in rice (Oryza sativa L.). Euphytica 2017, 213, 127. [Google Scholar] [CrossRef]
- Miura, K.; Lin, S.; Yano, M.; Nagamine, T. Mapping quantitative trait loci controlling seed longevity in rice (Oryza sativa L.). Theor. Appl. Genet. 2002, 104, 981–986. [Google Scholar] [CrossRef]
- Sasaki, K.; Takeuchi, Y.; Miura, K.; Yamaguchi, T.; Ando, T.; Ebitani, T.; Higashitani, A.; Yamaya, T.; Yano, M.; Sato, T. Fine mapping of a major quantitative trait locus, qLG-9, that controls seed longevity in rice (Oryza sativa L.). Theor. Appl. Genet. 2015, 128, 769–778. [Google Scholar] [CrossRef]
- Li, L.; Lin, Q.; Liu, S.; Liu, X.; Wang, W.; Hang, N.T.; Liu, F.; Zhao, Z.; Jiang, L.; Wan, J. Identification of quantitative trait loci for seed storability in rice (Oryza sativa L.). Plant. Breed. 2012, 131, 739–743. [Google Scholar] [CrossRef]
- Wang, Y.; Mostafa, S.; Zeng, W.; Jin, B. Function and mechanism of jasmonic acid in plant responses to abiotic and biotic stresses. Int. J. Mol. Sci. 2021, 22, 8568. [Google Scholar] [CrossRef]
- Singh, P.; Mukhopadhyay, K. Comprehensive molecular dissection of TIFY Transcription factors reveal their dynamic responses to biotic and abiotic stress in whea (Triticum aestivum L.). Sci. Rep. 2021, 11, 9739. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Reads | Clean Base (bp) | Mapping Rate (%) | Q30 (%) | GC (%) | Average Depth (×) | Coverage at Least 1× (%) | Coverage at Least 5× (%) | |
|---|---|---|---|---|---|---|---|---|
| Dongxiang wild rice (DXWR) | 33,922,707 | 10,157,432,212 | 97.54 | 93.95 | 44.63 | 20 | 97.36 | 93.27 |
| F6 | 33,982,607 | 10,144,910,192 | 96.60 | 94.70 | 45.32 | 21 | 91.06 | 86.93 |
| Bulked-intolerant BIL samples (IB-bulk) | 33,710,561 | 10,092,480,184 | 95.82 | 94.03 | 44.58 | 21 | 94.59 | 88.27 |
| Bulked-tolerant BIL samples (TB-bulk) | 39,408,702 | 11,791,891,500 | 95.31 | 91.54 | 43.71 | 24 | 94.44 | 88.63 |
| Chromosome ID | Start | End | Size (MB) | Gene Number |
|---|---|---|---|---|
| Chr4 | 18,550,000 | 18,550,000 | 0.00 | 1 |
| Chr4 | 18,600,000 | 19,670,000 | 1.07 | 162 |
| Chr4 | 20,620,000 | 20,630,000 | 0.01 | 3 |
| Chr4 | 20,650,000 | 20,680,000 | 0.03 | 3 |
| Chr4 | 20,770,000 | 20,820,000 | 0.05 | 8 |
| Chr4 | 20,860,000 | 20,870,000 | 0.01 | 1 |
| Chr9 | 7,860,000 | 9,780,000 | 1.92 | 270 |
| Total | - | - | 3.09 | 448 |
| Annotated Databases | Gene Number | Nonsynonymous- Mutation Gene Number | Frameshift- Mutation Gene Number |
|---|---|---|---|
| NR | 431 | 265 | 78 |
| NT | 448 | 274 | 82 |
| TrEMBL | 433 | 267 | 78 |
| SwissProt | 194 | 116 | 28 |
| GO | 337 | 196 | 53 |
| KEGG | 64 | 32 | 4 |
| GOG | 97 | 61 | 15 |
| Total | 448 | 274 | 82 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, M.; Hu, B.; Fan, Y.; Ding, G.; Yang, W.; Chen, Y.; Chen, Y.; Xie, J.; Zhang, F. Identification, Analysis, and Confirmation of Seed Storability-Related Loci in Dongxiang Wild Rice (Oryza rufipogon Griff.). Genes 2021, 12, 1831. https://doi.org/10.3390/genes12111831
Zhao M, Hu B, Fan Y, Ding G, Yang W, Chen Y, Chen Y, Xie J, Zhang F. Identification, Analysis, and Confirmation of Seed Storability-Related Loci in Dongxiang Wild Rice (Oryza rufipogon Griff.). Genes. 2021; 12(11):1831. https://doi.org/10.3390/genes12111831
Chicago/Turabian StyleZhao, Minmin, Biaolin Hu, Yuanwei Fan, Gumu Ding, Wanling Yang, Yong Chen, Yanhong Chen, Jiankun Xie, and Fantao Zhang. 2021. "Identification, Analysis, and Confirmation of Seed Storability-Related Loci in Dongxiang Wild Rice (Oryza rufipogon Griff.)" Genes 12, no. 11: 1831. https://doi.org/10.3390/genes12111831
APA StyleZhao, M., Hu, B., Fan, Y., Ding, G., Yang, W., Chen, Y., Chen, Y., Xie, J., & Zhang, F. (2021). Identification, Analysis, and Confirmation of Seed Storability-Related Loci in Dongxiang Wild Rice (Oryza rufipogon Griff.). Genes, 12(11), 1831. https://doi.org/10.3390/genes12111831