
Dyrk1a from Gene Function in Development and Physiology to Dosage Correction across Life Span in Down Syndrome


Abstract
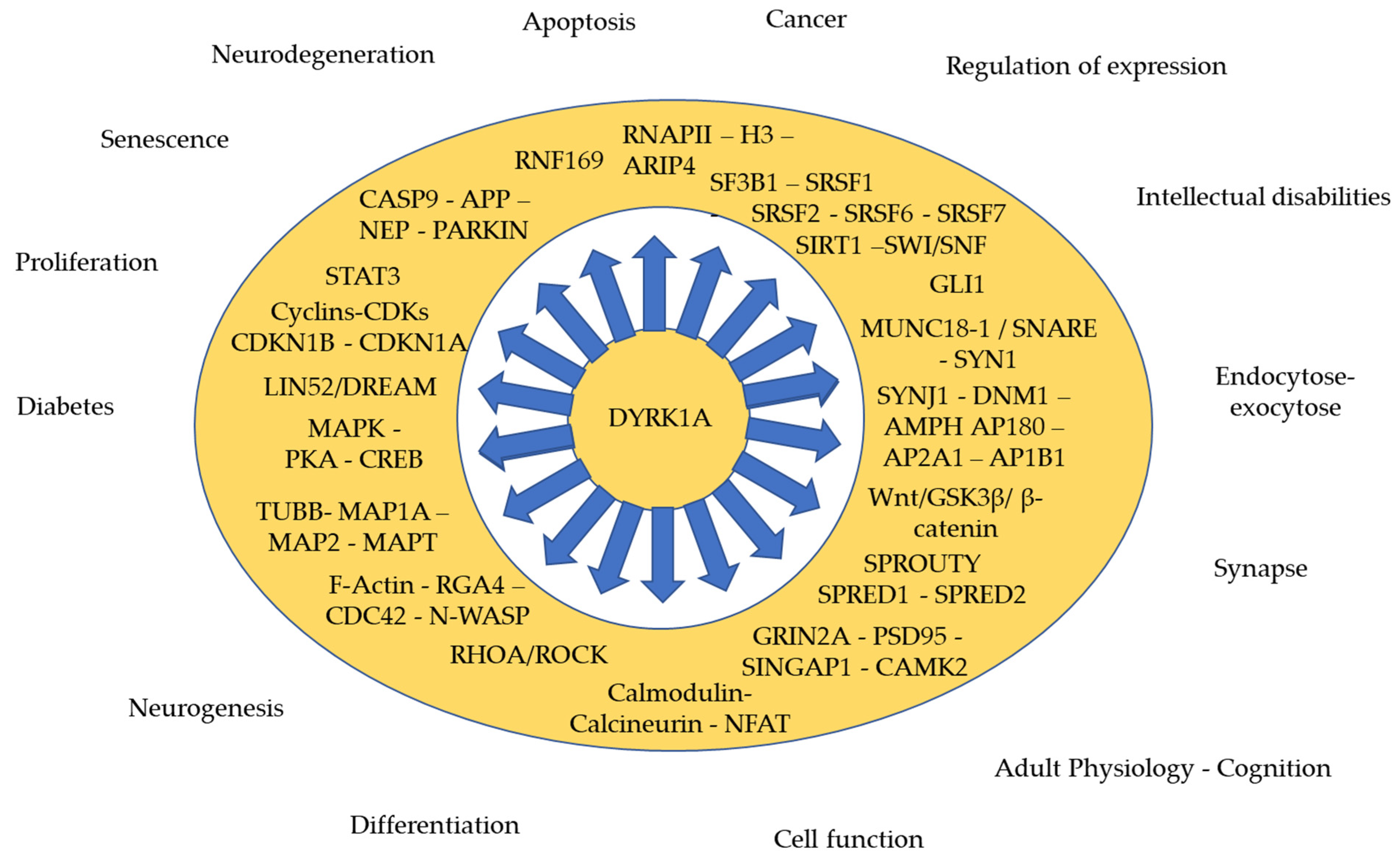
:1. Introduction
2. DYRK1A a Kinase with Multiple Roles at the Frontiers of Neuronal Proliferation, Differentiation, and Function
2.1. DYRK1A, a Unique Member of a Kinase Subfamily
2.1.1. The Family of DYRK Proteins
2.1.2. DYRK1A Expression and Expression Regulation
2.1.3. Regulation of DYRK1A Kinase Activity
2.2. Targets and Interactors of DYRK1A
2.2.1. During Cell Proliferation and Neurogenesis
2.2.2. Synaptic Function
2.2.3. Regulation of Expression (Methylation, Transcription, Translation)
2.2.4. Apoptosis, Neurodegeneration
3. DYRK1A as a Target for Improving DS Cognition in Young Adults
3.1. Natural Product Derived DYRK1A Inhibitors and Their Derivatives
3.1.1. Harmine, a Structural Analogue of β-Carbolin and an Indole Derivate
3.1.2. Epigallocatechin Gallate Is the Most Broadly Used DYRK1A Inhibitor
3.1.3. Imidazolone DYRK1A Inhibitor
3.2. Synthetic DYRK1A Inhibitors
3.3. Promising DYRK1A Inhibitors as Therapy for DS
3.3.1. Benzothiazole Derivates DYRK1A Inhibitors
3.3.2. Compounds Acting on DYRK1A Kinase Activity Stability
3.3.3. Compounds Discovered from Functional Cellular Analysis
4. Antenatal and Prenatal Treatment
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Varjosalo, M.; Keskitalo, S.; Van Drogen, A.; Nurkkala, H.; Vichalkovski, A.; Aebersold, R.; Gstaiger, M. The protein interaction landscape of the human CMGC kinase group. Cell Rep. 2013, 3, 1306–1320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrett, S.; Broach, J. Loss of Ras activity in Saccharomyces cerevisiae is suppressed by disruptions of a new kinase gene, YAKI, whose product may act downstream of the cAMP-dependent protein kinase. Genes Dev. 1989, 3, 1336–1348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejedor, F.; Zhu, X.R.; Kaltenbach, E.; Ackermann, A.; Baumann, A.; Canal, I.; Heisenberg, M.; Fischbach, K.F.; Pongs, O. minibrain: A new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron 1995, 14, 287–301. [Google Scholar] [CrossRef] [Green Version]
- Kinstrie, R.; Luebbering, N.; Miranda-Saavedra, D.; Sibbet, G.; Han, J.; Lochhead, P.A.; Cleghon, V. Characterization of a domain that transiently converts class 2 DYRKs into intramolecular tyrosine kinases. Sci. Signal. 2010, 3, ra16. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, M.; Estivill, X.; de la Luna, S. DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J. Cell Sci. 2003, 116, 3099–3107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexeeva, M.; Åberg, E.; Engh, R.A.; Rothweiler, U. The structure of a dual-specificity tyrosine phosphorylation-regulated kinase 1A-PKC412 complex reveals disulfide-bridge formation with the anomalous catalytic loop HRD(HCD) cysteine. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 1207–1215. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.; Chen, Y.; Chen, Z.; Dominique, R.; Glenn, K.; He, Y.; Janson, C.; Luk, K.C.; Lukacs, C.; Polonskaia, A.; et al. Pyrido[2,3-d]pyrimidines: Discovery and preliminary SAR of a novel series of DYRK1B and DYRK1A inhibitors. Bioorgan. Med. Chem. Lett. 2013, 23, 6610–6615. [Google Scholar] [CrossRef]
- Falke, H.; Chaikuad, A.; Becker, A.; Loaëc, N.; Lozach, O.; Abu Jhaisha, S.; Becker, W.; Jones, P.G.; Preu, L.; Baumann, K.; et al. 10-iodo-11H-indolo[3,2-c]quinoline-6-carboxylic acids are selective inhibitors of DYRK1A. J. Med. Chem. 2015, 58, 3131–3143. [Google Scholar] [CrossRef]
- Ogawa, Y.; Nonaka, Y.; Goto, T.; Ohnishi, E.; Hiramatsu, T.; Kii, I.; Yoshida, M.; Ikura, T.; Onogi, H.; Shibuya, H.; et al. Development of a novel selective inhibitor of the Down syndrome-related kinase Dyrk1A. Nat. Commun. 2010, 1, 86. [Google Scholar] [CrossRef] [Green Version]
- Tahtouh, T.; Elkins, J.M.; Filippakopoulos, P.; Soundararajan, M.; Burgy, G.; Durieu, E.; Cochet, C.; Schmid, R.S.; Lo, D.C.; Delhommel, F.; et al. Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J. Med. Chem. 2012, 55, 9312–9330. [Google Scholar] [CrossRef]
- Soundararajan, M.; Roos, A.K.; Savitsky, P.; Filippakopoulos, P.; Kettenbach, A.N.; Olsen, J.V.; Gerber, S.A.; Eswaran, J.; Knapp, S.; Elkins, J.M. Structures of Down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure 2013, 21, 986–996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothweiler, U.; Stensen, W.; Brandsdal, B.O.; Isaksson, J.; Leeson, F.A.; Engh, R.A.; Svendsen, J.S. Probing the ATP-Binding Pocket of Protein Kinase DYRK1A with Benzothiazole Fragment Molecules. J. Med. Chem. 2016, 59, 9814–9824. [Google Scholar] [CrossRef] [PubMed]
- Okui, M.; Ide, T.; Morita, K.; Funakoshi, E.; Ito, F.; Ogita, K.; Yoneda, Y.; Kudoh, J.; Shimizu, N. High-level expression of the Mnb/Dyrk1A gene in brain and heart during rat early development. Genomics 1999, 62, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, Z.; Lopes, C.; Rachidi, M.; Delabar, J.M. Expression of the mnb (dyrk) protein in adult and embryonic mouse tissues. Biochem. Biophys. Res. Commun. 1998, 253, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Guimera, J.; Casas, C.; Estivill, X.; Pritchard, M. Human minibrain homologue (MNBH/DYRK1): Characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics 1999, 57, 407–418. [Google Scholar] [CrossRef]
- Guimerá, J.; Casas, C.; Pucharcòs, C.; Solans, A.; Domènech, A.; Planas, A.M.; Ashley, J.; Lovett, M.; Estivill, X.; Pritchard, M.A. A human homologue of Drosophila minibrain (MNB) is expressed in the neuronal regions affected in Down syndrome and maps to the critical region. Hum. Meol. Gnet. 1996, 5, 1305–1310. [Google Scholar] [CrossRef] [Green Version]
- van Bon, B.W.; Coe, B.P.; Bernier, R.; Green, C.; Gerdts, J.; Witherspoon, K.; Kleefstra, T.; Willemsen, M.H.; Kumar, R.; Bosco, P.; et al. Disruptive de novo mutations of DYRK1A lead to a syndromic form of autism and ID. Mol. Psychiatry 2016, 21, 126–132. [Google Scholar] [CrossRef]
- Hämmerle, B.; Elizalde, C.; Tejedor, F.J. The spatio-temporal and subcellular expression of the candidate Down syndrome gene Mnb/Dyrk1A in the developing mouse brain suggests distinct sequential roles in neuronal development. Eur. J. Neurosci. 2008, 27, 1061–1074. [Google Scholar] [CrossRef]
- Hammerle, B.; Carnicero, A.; Elizalde, C.; Ceron, J.; Martinez, S.; Tejedor, F.J. Expression patterns and subcellular localization of the Down syndrome candidate protein MNB/DYRK1A suggest a role in late neuronal differentiation. Eur. J. Neurosci. 2003, 17, 2277–2286. [Google Scholar] [CrossRef]
- Marti, E.; Altafaj, X.; Dierssen, M.; de la Luna, S.; Fotaki, V.; Alvarez, M.; Perez-Riba, M.; Ferrer, I.; Estivill, X. Dyrk1A expression pattern supports specific roles of this kinase in the adult central nervous system. Brain Res. 2003, 964, 250–263. [Google Scholar] [CrossRef]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Dowjat, K.; Silverman, W.P.; Reisberg, B.; DeLeon, M.; Wisniewski, T.; Adayev, T.; et al. Cell type- and brain structure-specific patterns of distribution of minibrain kinase in human brain. Brain Res. 2004, 1010, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Maenz, B.; Hekerman, P.; Vela, E.M.; Galceran, J.; Becker, W. Characterization of the human DYRK1A promoter and its regulation by the transcription factor E2F1. BMC Mol. Biol. 2008, 9, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Zheng, L.; Han, B.; Wang, L.; Wang, P.; Liu, H.; Sun, X. REST Regulates DYRK1A Transcription in a Negative Feedback Loop. J. Biol. Chem. 2011, 286, 10755–10763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, W.; Zhou, H.; Liu, L.; Zhao, C.; Deng, Y.; Chen, L.; Wu, L.; Mandrycky, N.; McNabb, C.T.; Peng, Y.; et al. Disruption of neurogenesis and cortical development in transgenic mice misexpressing Olig2, a gene in the Down syndrome critical region. Neurobiol. Dis. 2015, 77, 106–116. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, L.; Best, T.K.; Cramer, N.P.; Carney, R.S.E.; Isaac, J.T.R.; Galdzicki, Z.; Haydar, T.F. Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat. Neurosci. 2010, 13, 927–934. [Google Scholar] [CrossRef] [Green Version]
- Vidaki, M.; Drees, F.; Saxena, T.; Lanslots, E.; Taliaferro, M.J.; Tatarakis, A.; Burge, C.B.; Wang, E.T.; Gertler, F.B. A Requirement for Mena, an Actin Regulator, in Local mRNA Translation in Developing Neurons. Neuron 2017, 95, 608–622. [Google Scholar] [CrossRef]
- Maier, B.; Gluba, W.; Bernier, B.; Turner, T.; Mohammad, K.; Guise, T.; Sutherland, A.; Thorner, M.; Scrable, H. Modulation of mammalian life span by the short isoform of p53. Genes Dev. 2004, 18, 306–319. [Google Scholar] [CrossRef] [Green Version]
- Pehar, M.; O’Riordan, K.J.; Burns-Cusato, M.; Andrzejewski, M.E.; del Alcazar, C.G.; Burger, C.; Scrable, H.; Puglielli, L. Altered longevity-assurance activity of p53:p44 in the mouse causes memory loss, neurodegeneration and premature death. Aging Cell 2010, 9, 174–190. [Google Scholar] [CrossRef] [Green Version]
- Pehar, M.; Ko, M.H.; Li, M.; Scrable, H.; Puglielli, L. P44, the ‘longevity-assurance’ isoform of P53, regulates tau phosphorylation and is activated in an age-dependent fashion. Aging Cell 2014, 13, 449–456. [Google Scholar] [CrossRef]
- Park, J.; Oh, Y.; Yoo, L.; Jung, M.S.; Song, W.J.; Lee, S.H.; Seo, H.; Chung, K.C. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J. Biol. Chem. 2010, 285, 31895–31906. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Liao, J.M.; Zeng, S.X.; Lu, H. p53 downregulates Down syndrome-associated DYRK1A through miR-1246. EMBO Rep. 2011, 12, 811–817. [Google Scholar] [CrossRef] [Green Version]
- da Costa Martins, P.A.; Salic, K.; Gladka, M.M.; Armand, A.S.; Leptidis, S.; el Azzouzi, H.; Hansen, A.; Coenen-de Roo, C.J.; Bierhuizen, M.F.; van der Nagel, R.; et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol. 2010, 12, 1220–1227. [Google Scholar] [CrossRef]
- Chiu, C.C.; Yeh, T.H.; Chen, R.S.; Chen, H.C.; Huang, Y.Z.; Weng, Y.H.; Cheng, Y.C.; Liu, Y.C.; Cheng, A.J.; Lu, Y.C.; et al. Upregulated Expression of MicroRNA-204-5p Leads to the Death of Dopaminergic Cells by Targeting DYRK1A-Mediated Apoptotic Signaling Cascade. Front. Cell Neurosci. 2019, 13, 399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göckler, N.; Jofre, G.; Papadopoulos, C.; Soppa, U.; Tejedor, F.J.; Becker, W. Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J. 2009, 276, 6324–6337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himpel, S.; Panzer, P.; Eirmbter, K.; Czajkowska, H.; Sayed, M.; Packman, L.C.; Blundell, T.; Kentrup, H.; Grötzinger, J.; Joost, H.G.; et al. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem. J. 2001, 359, 497–505. [Google Scholar] [CrossRef] [PubMed]
- Adayev, T.; Chen-Hwang, M.C.; Murakami, N.; Lee, E.; Bolton, D.C.; Hwang, Y.W. Dual-specificity tyrosine phosphorylation-regulated kinase 1A does not require tyrosine phosphorylation for activity in vitro. Biochemistry 2007, 46, 7614–7624. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Sibbet, G.; Morrice, N.; Cleghon, V. Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 2005, 121, 925–936. [Google Scholar] [CrossRef] [Green Version]
- Walte, A.; Rüben, K.; Birner-Gruenberger, R.; Preisinger, C.; Bamberg-Lemper, S.; Hilz, N.; Bracher, F.; Becker, W. Mechanism of dual specificity kinase activity of DYRK1A. FEBS J. 2013, 280, 4495–4511. [Google Scholar] [CrossRef]
- Kii, I.; Sumida, Y.; Goto, T.; Sonamoto, R.; Okuno, Y.; Yoshida, S.; Kato-Sumida, T.; Koike, Y.; Abe, M.; Nonaka, Y.; et al. Selective inhibition of the kinase DYRK1A by targeting its folding process. Nat. Commun. 2016, 7, 11391. [Google Scholar] [CrossRef]
- Alvarez, M.; Altafaj, X.; Aranda, S.; de la Luna, S. DYRK1A autophosphorylation on serine residue 520 modulates its kinase activity via 14-3-3 binding. Mol. Biol. Cell 2007, 18, 1167–1178. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Song, W.J.; Chung, K.C. Function and regulation of Dyrk1A: Towards understanding Down syndrome. Cell Mol. Life Sci. 2009, 66, 3235–3240. [Google Scholar] [CrossRef]
- Kida, E.; Walus, M.; Jarząbek, K.; Palminiello, S.; Albertini, G.; Rabe, A.; Hwang, Y.W.; Golabek, A.A. Form of dual-specificity tyrosine-(Y)-phosphorylation-regulated kinase 1A nonphosphorylated at tyrosine 145 and 147 is enriched in the nuclei of astroglial cells, adult hippocampal progenitors, and some cholinergic axon terminals. Neuroscience 2011, 195, 112–127. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Jackson, R.A.; Yusoff, P.; Guy, G.R. Direct association of Sprouty-related protein with an EVH1 domain (SPRED) 1 or SPRED2 with DYRK1A modifies substrate/kinase interactions. J. Biol. Chem. 2010, 285, 35374–35385. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.J.; Ahn, Y.S.; Chung, K.C. Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J. Biol. Chem. 2001, 276, 39819–39824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschöp, K.; Conery, A.R.; Litovchick, L.; Decaprio, J.A.; Settleman, J.; Harlow, E.; Dyson, N. A kinase shRNA screen links LATS2 and the pRB tumor suppressor. Genes Dev. 2011, 25, 814–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Im, E.; Hyun, M.; Park, J.; Chung, K.C. Protein phosphatase PPM1B inhibits DYRK1A kinase through dephosphorylation of pS258 and reduces toxic tau aggregation. J. Biol. Chem. 2021, 296, 100245. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Yin, X.; Gu, J.; Zhang, X.; Shi, J.; Qian, W.; Ji, Y.; Cao, M.; Gu, X.; Ding, F.; et al. Truncation and Activation of Dual Specificity Tyrosine Phosphorylation-regulated Kinase 1A by Calpain I: A molecular mechanism linked to tau pathology in Alzheimer disease. J. Biol. Chem. 2015, 290, 15219–15237. [Google Scholar] [CrossRef] [Green Version]
- Souchet, B.; Audrain, M.; Billard, J.M.; Dairou, J.; Fol, R.; Orefice, N.S.; Tada, S.; Gu, Y.; Dufayet-Chaffaud, G.; Limanton, E.; et al. Inhibition of DYRK1A proteolysis modifies its kinase specificity and rescues Alzheimer phenotype in APP/PS1 mice. Acta Neuropathol. Commun. 2019, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Tang, Y.; Chen, L.; Liu, N.; Lang, F.; Liu, H.; Wang, P.; Sun, X. E3 Ligase SCFβTrCP-induced DYRK1A Protein Degradation Is Essential for Cell Cycle Progression in HEK293 Cells. J. Biol. Chem. 2016, 291, 26399–26409. [Google Scholar] [CrossRef] [Green Version]
- Becker, W.; Weber, Y.; Wetzel, K.; Eirmbter, K.; Tejedor, F.J.; Joost, H.G. Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J. Biol. Chem. 1998, 273, 25893–25902. [Google Scholar] [CrossRef] [Green Version]
- Aranda, S.; Alvarez, M.; Turró, S.; Laguna, A.; de la Luna, S. Sprouty2-mediated inhibition of fibroblast growth factor signaling is modulated by the protein kinase DYRK1A. Mol. Cell Biol. 2008, 28, 5899–5911. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.L.; Duchon, A.; Manousopoulou, A.; Loaëc, N.; Villiers, B.; Pani, G.; Karatas, M.; Mechling, A.E.; Harsan, L.A.; Limanton, E.; et al. Correction of cognitive deficits in mouse models of Down syndrome by a pharmacological inhibitor of DYRK1A. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Shibata, T.; Aoshima, M.; Tsubata, T.; Nishida, E. The molecular chaperone TRiC/CCT binds to the Trp-Asp 40 (WD40) repeat protein WDR68 and promotes its folding, protein kinase DYRK1A binding, and nuclear accumulation. J. Biol. Chem. 2014, 289, 33320–33332. [Google Scholar] [CrossRef] [Green Version]
- Miyata, Y.; Nishida, E. DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation. Biochim. Biophys. Acta 2011, 1813, 1728–1739. [Google Scholar] [CrossRef] [Green Version]
- Bescond, M.; Rahmani, Z. Dual-specificity tyrosine-phosphorylated and regulated kinase 1A (DYRK1A) interacts with the phytanoyl-CoA alpha-hydroxylase associated protein 1 (PAHX-AP1), a brain specific protein. Int. J. Biochem. Cell Biol. 2005, 37, 775–783. [Google Scholar] [CrossRef]
- Kaczmarski, W.; Barua, M.; Mazur-Kolecka, B.; Frackowiak, J.; Dowjat, W.; Mehta, P.; Bolton, D.; Hwang, Y.W.; Rabe, A.; Albertini, G.; et al. Intracellular distribution of differentially phosphorylated dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A). J. Neurosci. Res. 2014, 92, 162–173. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.J.; Zhu, Y.W.; Zhang, J.L.; Cheng, J.F.; Rubin, E.M. Construction of a panel of transgenic mice containing a contiguous 2-Mb set of YAC/P1 clones form human chromosome 21q22.2. Genomics 1995, 27, 425–434. [Google Scholar] [CrossRef]
- Altafaj, X.; Dierssen, M.; Baamonde, C.; Marti, E.; Visa, J.; Guimera, J.; Oset, M.; Gonzalez, J.R.; Florez, J.; Fillat, C.; et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum. Mol. Genet. 2001, 10, 1915–1923. [Google Scholar] [CrossRef] [Green Version]
- Sago, H.; Carlson, E.J.; Smith, D.J.; Kilbridge, J.; Rubin, E.M.; Mobley, W.C.; Epstein, C.J.; Huang, T.T. Ts1Cje, a partial trisomy 16 mouse model for Down syndrome, exhibits learning and behavioral abnormalities. Proc. Natl. Acad. Sci. USA 1998, 95, 6256–6261. [Google Scholar] [CrossRef] [Green Version]
- Arranz, J.; Balducci, E.; Arató, K.; Sánchez-Elexpuru, G.; Najas, S.; Parras, A.; Rebollo, E.; Pijuan, I.; Erb, I.; Verde, G.; et al. Impaired development of neocortical circuits contributes to the neurological alterations in DYRK1A haploinsufficiency syndrome. Neurobiol. Dis. 2019, 127, 210–222. [Google Scholar] [CrossRef]
- Sitz, J.H.; Tigges, M.; Baumgärtel, K.; Khaspekov, L.G.; Lutz, B. Dyrk1A potentiates steroid hormone-induced transcription via the chromatin remodeling factor Arip4. Mol. Cell Biol. 2004, 24, 5821–5834. [Google Scholar] [CrossRef] [Green Version]
- Kung, J.E.; Jura, N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 2016, 24, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Himpel, S.; Tegge, W.; Frank, R.; Leder, S.; Joost, H.G.; Becker, W. Specificity determinants of substrate recognition by the protein kinase DYRK1A. J. Biol. Chem. 2000, 275, 2431–2438. [Google Scholar] [CrossRef] [Green Version]
- Ryoo, S.R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.J.; Cho, H.J.; Lee, H.W.; Kim, I.S.; Cheon, Y.H.; Ahn, Y.S.; Chung, S.H.; et al. DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Liang, Z.; Wegiel, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; Ramakrishna, N.; Gong, C.X. Overexpression of Dyrk1A contributes to neurofibrillary degeneration in Down syndrome. FASEB J. 2008, 22, 3224–3233. [Google Scholar] [CrossRef] [Green Version]
- Murakami, N.; Xie, W.; Lu, R.C.; Chen-Hwang, M.C.; Wieraszko, A.; Hwang, Y.W. Phosphorylation of amphiphysin I by minibrain kinase/dual-specificity tyrosine phosphorylation-regulated kinase, a kinase implicated in Down syndrome. J. Biol. Chem. 2006, 281, 23712–23724. [Google Scholar] [CrossRef] [Green Version]
- Seifert, A.; Clarke, P.R. p38alpha- and DYRK1A-dependent phosphorylation of caspase-9 at an inhibitory site in response to hyperosmotic stress. Cell Signal. 2009, 21, 1626–1633. [Google Scholar] [CrossRef]
- de Graaf, K.; Czajkowska, H.; Rottmann, S.; Packman, L.C.; Lilischkis, R.; Lüscher, B.; Becker, W. The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochem. 2006, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.J.; Sung, J.Y.; Lee, H.J.; Rhim, H.; Hasegawa, M.; Iwatsubo, T.; Min, d.S.; Kim, J.; Paik, S.R.; Chung, K.C. Dyrk1A phosphorylates alpha-synuclein and enhances intracellular inclusion formation. J. Biol. Chem. 2006, 281, 33250–33257. [Google Scholar] [CrossRef] [Green Version]
- Becker, W. Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle 2012, 11, 3389–3394. [Google Scholar] [CrossRef] [Green Version]
- Møller, R.S.; Kübart, S.; Hoeltzenbein, M.; Heye, B.; Vogel, I.; Hansen, C.P.; Menzel, C.; Ullmann, R.; Tommerup, N.; Ropers, H.H.; et al. Truncation of the Down syndrome candidate gene DYRK1A in two unrelated patients with microcephaly. Am. J. Hum. Genet. 2008, 82, 1165–1170. [Google Scholar] [CrossRef] [Green Version]
- Courcet, J.B.; Faivre, L.; Malzac, P.; Masurel-Paulet, A.; Lopez, E.; Callier, P.; Lambert, L.; Lemesle, M.; Thevenon, J.; Gigot, N.; et al. The DYRK1A gene is a cause of syndromic intellectual disability with severe microcephaly and epilepsy. J. Med. Genet. 2012, 49, 731–736. [Google Scholar] [CrossRef]
- van Bon, B.W.; Hoischen, A.; Hehir-Kwa, J.; de Brouwer, A.P.; Ruivenkamp, C.; Gijsbers, A.C.; Marcelis, C.L.; de Leeuw, N.; Veltman, J.A.; Brunner, H.G.; et al. Intragenic deletion in DYRK1A leads to mental retardation and primary microcephaly. Clin. Genet. 2011, 79, 296–299. [Google Scholar] [CrossRef]
- Fotaki, V.; Dierssen, M.; Alcantara, S.; Martinez, S.; Marti, E.; Casas, C.; Visa, J.; Soriano, E.; Estivill, X.; Arbones, M.L. Dyrk1A haploinsufficiency affects viability and causes developmental delay and abnormal brain morphology in mice. Mol. Cell Biol. 2002, 22, 6636–6647. [Google Scholar] [CrossRef] [Green Version]
- Tejedor, F.J.; Hämmerle, B. MNB/DYRK1A as a multiple regulator of neuronal development. FEBS J. 2011, 278, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Yabut, O.; Domogauer, J.; D’Arcangelo, G. Dyrk1A overexpression inhibits proliferation and induces premature neuronal differentiation of neural progenitor cells. J. Neurosci. 2010, 30, 4004–4014. [Google Scholar] [CrossRef]
- Contestabile, A.; Fila, T.; Ceccarelli, C.; Bonasoni, P.; Bonapace, L.; Santini, D.; Bartesaghi, R.; Ciani, E. Cell cycle alteration and decreased cell proliferation in the hippocampal dentate gyrus and in the neocortical germinal matrix of fetuses with Down syndrome and in Ts65Dn mice. Hippocampus 2007, 17, 665–678. [Google Scholar] [CrossRef]
- Larsen, K.B.; Laursen, H.; Graem, N.; Samuelsen, G.B.; Bogdanovic, N.; Pakkenberg, B. Reduced cell number in the neocortical part of the human fetal brain in Down syndrome. Ann. Anat. 2008, 190, 421–427. [Google Scholar] [CrossRef]
- Hämmerle, B.; Ulin, E.; Guimera, J.; Becker, W.; Guillemot, F.; Tejedor, F.J. Transient expression of Mnb/Dyrk1a couples cell cycle exit and differentiation of neuronal precursors by inducing p27KIP1 expression and suppressing NOTCH signaling. Development 2011, 138, 2543–2554. [Google Scholar] [CrossRef] [Green Version]
- Soppa, U.; Schumacher, J.; Florencio Ortiz, V.; Pasqualon, T.; Tejedor, F.J.; Becker, W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle 2014, 13, 2084–2100. [Google Scholar] [CrossRef] [Green Version]
- Shaikh, M.N.; Gutierrez-Avino, F.; Colonques, J.; Ceron, J.; Hammerle, B.; Tejedor, F.J. Minibrain drives the Dacapo-dependent cell cycle exit of neurons in the Drosophila brain by promoting asense and prospero expression. Development 2016, 143, 3195–3205. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.Y.; Lin, J.R.; Tsai, F.C.; Meyer, T. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol. Cell 2013, 52, 87–100. [Google Scholar] [CrossRef] [Green Version]
- Litovchick, L.; Florens, L.A.; Swanson, S.K.; Washburn, M.P.; DeCaprio, J.A. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011, 25, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Levy, J.A.; LaFlamme, C.W.; Tsaprailis, G.; Crynen, G.; Page, D.T. Dyrk1a Mutations Cause Undergrowth of Cortical Pyramidal Neurons via Dysregulated Growth Factor Signaling. Biol. Psychiatry 2021, 90, 295–306. [Google Scholar] [CrossRef]
- Najas, S.; Arranz, J.; Lochhead, P.A.; Ashford, A.L.; Oxley, D.; Delabar, J.M.; Cook, S.J.; Barallobre, M.J.; Arbonés, M.L. DYRK1A-mediated Cyclin D1 Degradation in Neural Stem Cells Contributes to the Neurogenic Cortical Defects in Down Syndrome. EBioMedicine 2015, 2, 120–134. [Google Scholar] [CrossRef] [Green Version]
- de Graaf, K.; Hekerman, P.; Spelten, O.; Herrmann, A.; Packman, L.C.; Büssow, K.; Müller-Newen, G.; Becker, W. Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: Phosphorylation by DYRK1A and colocalization with splicing factors. J. Biol. Chem. 2004, 279, 4612–4624. [Google Scholar] [CrossRef] [Green Version]
- Kinstrie, R.; Lochhead, P.A.; Sibbet, G.; Morrice, N.; Cleghon, V. dDYRK2 and Minibrain interact with the chromatin remodelling factors SNR1 and TRX. Biochem. J. 2006, 398, 45–54. [Google Scholar] [CrossRef]
- Kelly, P.A.; Rahmani, Z. DYRK1A enhances the mitogen-activated protein kinase cascade in PC12 cells by forming a complex with Ras, B-Raf, and MEK1. Mol. Biol. Cell 2005, 16, 3562–3573. [Google Scholar] [CrossRef] [Green Version]
- Mao, J.; Maye, P.; Kogerman, P.; Tejedor, F.J.; Toftgard, R.; Xie, W.; Wu, G.; Wu, D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J. Biol. Chem. 2002, 277, 35156–35161. [Google Scholar] [CrossRef] [Green Version]
- Ehe, B.K.; Lamson, D.R.; Tarpley, M.; Onyenwoke, R.U.; Graves, L.M.; Williams, K.P. Identification of a DYRK1A-mediated phosphorylation site within the nuclear localization sequence of the hedgehog transcription factor GLI1. Biochem. Biophys. Res. Commun. 2017, 491, 767–772. [Google Scholar] [CrossRef]
- Fernandez-Martinez, J.; Vela, E.M.; Tora-Ponsioen, M.; Ocaña, O.H.; Nieto, M.A.; Galceran, J. Attenuation of Notch signalling by the Down-syndrome-associated kinase DYRK1A. J. Cell Sci. 2009, 122, 1574–1583. [Google Scholar] [CrossRef] [Green Version]
- Skurat, A.V.; Dietrich, A.D. Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J. Biol. Chem. 2004, 279, 2490–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, Y.L.; Rena, G.; Morrice, N.; Barthel, A.; Becker, W.; Guo, S.; Unterman, T.G.; Cohen, P. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem. J. 2001, 355, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Scales, T.M.; Lin, S.; Kraus, M.; Goold, R.G.; Gordon-Weeks, P.R. Nonprimed and DYRK1A-primed GSK3 beta-phosphorylation sites on MAP1B regulate microtubule dynamics in growing axons. J. Cell Sci. 2009, 122, 2424–2435. [Google Scholar] [CrossRef] [Green Version]
- Tatebe, H.; Nakano, K.; Maximo, R.; Shiozaki, K. Pom1 DYRK regulates localization of the Rga4 GAP to ensure bipolar activation of Cdc42 in fission yeast. Curr. Biol. 2008, 18, 322–330. [Google Scholar] [CrossRef] [Green Version]
- Park, J.; Sung, J.Y.; Song, W.J.; Chang, S.; Chung, K.C. Dyrk1A negatively regulates the actin cytoskeleton through threonine phosphorylation of N-WASP. J. Cell Sci. 2012, 125, 67–80. [Google Scholar] [CrossRef] [Green Version]
- Abekhoukh, S.; Planque, C.; Ripoll, C.; Urbaniak, P.; Paul, J.L.; Delabar, J.M.; Janel, N. Dyrk1A, a serine/threonine kinase, is involved in ERK and Akt activation in the brain of hyperhomocysteinemic mice. Mol. Neurobiol. 2013, 47, 105–116. [Google Scholar] [CrossRef]
- Shin, J.H.; Guedj, F.; Delabar, J.M.; Lubec, G. Dysregulation of growth factor receptor-bound protein 2 and fascin in hippocampus of mice polytransgenic for chromosome 21 structures. Hippocampus 2007, 17, 1180–1192. [Google Scholar] [CrossRef]
- Park, J.; Oh, Y.; Chung, K.C. Two key genes closely implicated with the neuropathological characteristics in Down syndrome: DYRK1A and RCAN1. BMB Rep. 2009, 42, 6–15. [Google Scholar] [CrossRef] [Green Version]
- Gwack, Y.; Sharma, S.; Nardone, J.; Tanasa, B.; Iuga, A.; Srikanth, S.; Okamura, H.; Bolton, D.; Feske, S.; Hogan, P.G.; et al. A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 2006, 441, 646–650. [Google Scholar] [CrossRef]
- Arron, J.R.; Winslow, M.M.; Polleri, A.; Chang, C.P.; Wu, H.; Gao, X.; Neilson, J.R.; Chen, L.; Heit, J.J.; Kim, S.K.; et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 2006, 441, 595–600. [Google Scholar] [CrossRef]
- Lee, Y.; Ha, J.; Kim, H.J.; Kim, Y.S.; Chang, E.J.; Song, W.J.; Kim, H.H. Negative feedback Inhibition of NFATc1 by DYRK1A regulates bone homeostasis. J. Biol. Chem. 2009, 284, 33343–33351. [Google Scholar] [CrossRef] [Green Version]
- Song, W.J.; Song, E.A.; Choi, S.H.; Baik, H.H.; Jin, B.K.; Kim, J.H.; Chung, S.H. Dyrk1A-mediated phosphorylation of RCAN1 promotes the formation of insoluble RCAN1 aggregates. Neurosci. Lett. 2013, 554, 135–140. [Google Scholar] [CrossRef]
- Jung, M.S.; Park, J.H.; Ryu, Y.S.; Choi, S.H.; Yoon, S.H.; Kwen, M.Y.; Oh, J.Y.; Song, W.J.; Chung, S.H. Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J. Biol. Chem. 2011, 286, 40401–40412. [Google Scholar] [CrossRef] [Green Version]
- Di Vona, C.; Bezdan, D.; Islam, A.B.; Salichs, E.; López-Bigas, N.; Ossowski, S.; de la Luna, S. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol. Cell 2015, 57, 506–520. [Google Scholar] [CrossRef] [Green Version]
- Chen-Hwang, M.C.; Chen, H.R.; Elzinga, M.; Hwang, Y.W. Dynamin is a minibrain kinase/dual specificity Yak1-related kinase 1A substrate. J. Biol. Chem. 2002, 277, 17597–17604. [Google Scholar] [CrossRef] [Green Version]
- Murakami, N.; Bolton, D.C.; Kida, E.; Xie, W.; Hwang, Y.W. Phosphorylation by Dyrk1A of clathrin coated vesicle-associated proteins: Identification of the substrate proteins and the effects of phosphorylation. PLoS ONE 2012, 7, e34845. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Chen-Hwang, M.C.; Dolios, G.; Murakami, N.; Padovan, J.C.; Wang, R.; Hwang, Y.W. Mnb/Dyrk1A phosphorylation regulates the interaction of dynamin 1 with SH3 domain-containing proteins. Biochemistry 2004, 43, 10173–10185. [Google Scholar] [CrossRef]
- Murakami, N.; Bolton, D.; Hwang, Y.W. Dyrk1A binds to multiple endocytic proteins required for formation of clathrin-coated vesicles. Biochemistry 2009, 48, 9297–9305. [Google Scholar] [CrossRef]
- Adayev, T.; Chen-Hwang, M.C.; Murakami, N.; Wang, R.; Hwang, Y.W. MNB/DYRK1A phosphorylation regulates the interactions of synaptojanin 1 with endocytic accessory proteins. Biochem. Biophys. Res. Commun. 2006, 351, 1060–1065. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.K.; Bregere, C.; Paluch, J.; Lu, J.F.; Dickman, D.K.; Chang, K.T. Activity-dependent facilitation of Synaptojanin and synaptic vesicle recycling by the Minibrain kinase. Nat. Commun. 2014, 5, 4246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.J.; Song, E.A.; Jung, M.S.; Choi, S.H.; Baik, H.H.; Jin, B.K.; Kim, J.H.; Chung, S.H. Phosphorylation and inactivation of glycogen synthase kinase 3β (GSK3β) by dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A). J. Biol. Chem. 2015, 290, 2321–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ori-McKenney, K.M.; McKenney, R.J.; Huang, H.H.; Li, T.; Meltzer, S.; Jan, L.Y.; Vale, R.D.; Wiita, A.P.; Jan, Y.N. Phosphorylation of β-Tubulin by the Down Syndrome Kinase, Minibrain/DYRK1a, Regulates Microtubule Dynamics and Dendrite Morphogenesis. Neuron 2016, 90, 551–563. [Google Scholar] [CrossRef] [Green Version]
- Park, J.H.; Jung, M.S.; Kim, Y.S.; Song, W.J.; Chung, S.H. Phosphorylation of Munc18-1 by Dyrk1A regulates its interaction with Syntaxin 1 and X11α. J. Neurochem. 2012, 122, 1081–1091. [Google Scholar] [CrossRef]
- Altafaj, X.; Ortiz-Abalia, J.; Fernández, M.; Potier, M.C.; Laffaire, J.; Andreu, N.; Dierssen, M.; González-García, C.; Ceña, V.; Martí, E.; et al. Increased NR2A expression and prolonged decay of NMDA-induced calcium transient in cerebellum of TgDyrk1A mice, a mouse model of Down syndrome. Neurobiol. Dis. 2008, 32, 377–384. [Google Scholar] [CrossRef]
- Grau, C.; Arató, K.; Fernández-Fernández, J.M.; Valderrama, A.; Sindreu, C.; Fillat, C.; Ferrer, I.; de la Luna, S.; Altafaj, X. DYRK1A-mediated phosphorylation of GluN2A at Ser(1048) regulates the surface expression and channel activity of GluN1/GluN2A receptors. Front. Cell Neurosci. 2014, 8, 331. [Google Scholar] [CrossRef] [Green Version]
- Brault, V.; Nguyen, T.L.; Flores-Gutiérrez, J.; Iacono, G.; Birling, M.C.; Lalanne, V.; Meziane, H.; Manousopoulou, A.; Pavlovic, G.; Lindner, L.; et al. Dyrk1a gene dosage in glutamatergic neurons has key effects in cognitive deficits observed in mouse models of MRD7 and Down syndrome. PLoS Genet. 2021, 17, e1009777. [Google Scholar] [CrossRef]
- Souchet, B.; Guedj, F.; Sahun, I.; Duchon, A.; Daubigney, F.; Badel, A.; Yanagawa, Y.; Jose Barallobre, M.; Dierssen, M.; Yu, E.; et al. Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol. Dis. 2014, 69, 65–75. [Google Scholar] [CrossRef]
- Lepagnol-Bestel, A.; Zvara, A.; Maussion, G.; Quignon, F.; Ngimbous, B.; Ramoz, N.; Imbeaud, S.; Loe-Mie, Y.; Benihoud, K.; Agier, N.; et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum. Mol. Genet. 2009, 18, 1405–1414. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.M.; Azebi, S.; Soubigou, G.; Muchardt, C. DYRK1A phoshorylates histone H3 to differentially regulate the binding of HP1 isoforms and antagonize HP1-mediated transcriptional repression. EMBO Rep. 2014, 15, 686–694. [Google Scholar] [CrossRef]
- Shi, J.; Zhang, T.; Zhou, C.; Chohan, M.O.; Gu, X.; Wegiel, J.; Zhou, J.; Hwang, Y.W.; Iqbal, K.; Grundke-Iqbal, I.; et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J. Biol. Chem. 2008, 283, 28660–28669. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Shi, J.; Qian, W.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. Regulation of alternative splicing of tau exon 10 by 9G8 and Dyrk1A. Neurobiol. Aging 2012, 33, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Qian, W.; Liang, H.; Shi, J.; Jin, N.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X.; Liu, F. Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 2011, 39, 6161–6171. [Google Scholar] [CrossRef] [Green Version]
- Yin, X.; Jin, N.; Gu, J.; Shi, J.; Zhou, J.; Gong, C.X.; Iqbal, K.; Grundke-Iqbal, I.; Liu, F. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) modulates serine/arginine-rich protein 55 (SRp55)-promoted Tau exon 10 inclusion. J. Biol. Chem. 2012, 287, 30497–30506. [Google Scholar] [CrossRef] [Green Version]
- Guard, S.E.; Poss, Z.C.; Ebmeier, C.C.; Pagratis, M.; Simpson, H.; Taatjes, D.J.; Old, W.M. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci. Rep. 2019, 9, 6539. [Google Scholar] [CrossRef] [Green Version]
- Menon, V.R.; Ananthapadmanabhan, V.; Swanson, S.; Saini, S.; Sesay, F.; Yakovlev, V.; Florens, L.; DeCaprio, J.A.; Washburn, M.P.; Dozmorov, M.; et al. DYRK1A regulates the recruitment of 53BP1 to the sites of DNA damage in part through interaction with RNF169. Cell Cycle 2019, 18, 531–551. [Google Scholar] [CrossRef] [Green Version]
- Roewenstrunk, J.; Di Vona, C.; Chen, J.; Borras, E.; Dong, C.; Arató, K.; Sabidó, E.; Huen, M.S.Y.; de la Luna, S. A comprehensive proteomics-based interaction screen that links DYRK1A to RNF169 and to the DNA damage response. Sci. Rep. 2019, 9, 6014. [Google Scholar] [CrossRef]
- Woods, Y.L.; Cohen, P.; Becker, W.; Jakes, R.; Goedert, M.; Wang, X.; Proud, C.G. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: Potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem. J. 2001, 355, 609–615. [Google Scholar] [CrossRef]
- Park, J.; Yang, E.J.; Yoon, J.H.; Chung, K.C. Dyrk1A overexpression in immortalized hippocampal cells produces the neuropathological features of Down syndrome. Mol. Cell Neurosci. 2007, 36, 270–279. [Google Scholar] [CrossRef]
- Ryoo, S.R.; Cho, H.J.; Lee, H.W.; Jeong, H.K.; Radnaabazar, C.; Kim, Y.S.; Kim, M.J.; Son, M.Y.; Seo, H.; Chung, S.H.; et al. Dual-specificity tyrosine(Y)-phosphorylation regulated kinase 1A-mediated phosphorylation of amyloid precursor protein: Evidence for a functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2008, 104, 1333–1344. [Google Scholar] [CrossRef]
- Vingtdeux, V.; Hamdane, M.; Gompel, M.; Bégard, S.; Drobecq, H.; Ghestem, A.; Grosjean, M.E.; Kostanjevecki, V.; Grognet, P.; Vanmechelen, E.; et al. Phosphorylation of amyloid precursor carboxy-terminal fragments enhances their processing by a gamma-secretase-dependent mechanism. Neurobiol. Dis. 2005, 20, 625–637. [Google Scholar] [CrossRef] [PubMed]
- Seifert, A.; Allan, L.A.; Clarke, P.R. DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J. 2008, 275, 6268–6280. [Google Scholar] [CrossRef] [PubMed]
- Laguna, A.; Aranda, S.; Barallobre, M.J.; Barhoum, R.; Fernández, E.; Fotaki, V.; Delabar, J.M.; de la Luna, S.; de la Villa, P.; Arbonés, M.L. The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev. Cell 2008, 15, 841–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burré, J. The Synaptic Function of α-Synuclein. J. Parkinson’s Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Williams, J.G.; Schug, T.T.; Li, X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J. Biol. Chem. 2010, 285, 13223–13232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sitz, J.H.; Baumgärtel, K.; Hämmerle, B.; Papadopoulos, C.; Hekerman, P.; Tejedor, F.J.; Becker, W.; Lutz, B. The Down syndrome candidate dual-specificity tyrosine phosphorylation-regulated kinase 1A phosphorylates the neurodegeneration-related septin 4. Neuroscience 2008, 157, 596–605. [Google Scholar] [CrossRef]
- Ryu, Y.S.; Park, S.Y.; Jung, M.S.; Yoon, S.H.; Kwen, M.Y.; Lee, S.Y.; Choi, S.H.; Radnaabazar, C.; Kim, M.K.; Kim, H.; et al. Dyrk1A-mediated phosphorylation of Presenilin 1: A functional link between Down syndrome and Alzheimer’s disease. J. Neurochem. 2010, 115, 574–584. [Google Scholar] [CrossRef] [PubMed]
- Asai, M.; Kawakubo, T.; Mori, R.; Iwata, N. Elucidating Pathogenic Mechanisms of Early-onset Alzheimer’s Disease in Down Syndrome Patients. Yakugaku Zasshi 2017, 137, 801–805. [Google Scholar] [CrossRef] [Green Version]
- Kawakubo, T.; Mori, R.; Shirotani, K.; Iwata, N.; Asai, M. Neprilysin Is Suppressed by Dual-Specificity Tyrosine-Phosphorylation Regulated Kinase 1A (DYRK1A) in Down-Syndrome-Derived Fibroblasts. Biol. Pharm. Bull. 2017, 40, 327–333. [Google Scholar] [CrossRef] [Green Version]
- Alldred, M.J.; Chao, H.M.; Lee, S.H.; Beilin, J.; Powers, B.E.; Petkova, E.; Strupp, B.J.; Ginsberg, S.D. CA1 pyramidal neuron gene expression mosaics in the Ts65Dn murine model of Down syndrome and Alzheimer’s disease following maternal choline supplementation. Hippocampus 2018, 28, 251–268. [Google Scholar] [CrossRef]
- Im, E.; Chung, K.C. Dyrk1A phosphorylates parkin at Ser-131 and negatively regulates its ubiquitin E3 ligase activity. J. Neurochem. 2015, 134, 756–768. [Google Scholar] [CrossRef]
- Kang, J.E.; Choi, S.A.; Park, J.B.; Chung, K.C. Regulation of the proapoptotic activity of huntingtin interacting protein 1 by Dyrk1 and caspase-3 in hippocampal neuroprogenitor cells. J. Neurosci. Res. 2005, 81, 62–72. [Google Scholar] [CrossRef]
- Xiang, J.; Yang, S.; Xin, N.; Gaertig, M.A.; Reeves, R.H.; Li, S.; Li, X.J. DYRK1A regulates Hap1-Dcaf7/WDR68 binding with implication for delayed growth in Down syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E1224–E1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willsey, H.R.; Xu, Y.; Everitt, A.; Dea, J.; Exner, C.R.T.; Willsey, A.J.; State, M.W.; Harland, R.M. The neurodevelopmental disorder risk gene DYRK1A is required for ciliogenesis and control of brain size in Xenopus embryos. Development 2020, 147. [Google Scholar] [CrossRef]
- Suetsugu, M.; Mehraein, P. Spine distribution along the apical dendrites of the pyramidal neurons in Down’s syndrome. A quantitative Golgi study. Acta Neuropathol. 1980, 50, 207–210. [Google Scholar] [CrossRef]
- Ferrer, I.; Gullotta, F. Down’s syndrome and Alzheimer’s disease: Dendritic spine counts in the hippocampus. Acta Neuropathol. 1990, 79, 680–685. [Google Scholar] [CrossRef] [PubMed]
- Haas, M.A.; Bell, D.; Slender, A.; Lana-Elola, E.; Watson-Scales, S.; Fisher, E.M.; Tybulewicz, V.L.; Guillemot, F. Alterations to dendritic spine morphology, but not dendrite patterning, of cortical projection neurons in Tc1 and Ts1Rhr mouse models of Down syndrome. PLoS ONE 2013, 8, e78561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez de Lagran, M.; Benavides-Piccione, R.; Ballesteros-Yañez, I.; Calvo, M.; Morales, M.; Fillat, C.; Defelipe, J.; Ramakers, G.J.; Dierssen, M. Dyrk1A influences neuronal morphogenesis through regulation of cytoskeletal dynamics in mammalian cortical neurons. Cereb. Cortex 2012, 22, 2867–2877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benavides-Piccione, R.; Dierssen, M.; Ballesteros-Yanez, I.; Martinez de Lagran, M.; Arbones, M.L.; Fotaki, V.; DeFelipe, J.; Elston, G.N. Alterations in the phenotype of neocortical pyramidal cells in the Dyrk1A+/− mouse. Neurobiol. Dis. 2005, 20, 115–122. [Google Scholar] [CrossRef]
- Dang, T.; Duan, W.Y.; Yu, B.; Tong, D.L.; Cheng, C.; Zhang, Y.F.; Wu, W.; Ye, K.; Zhang, W.X.; Wu, M.; et al. Autism-associated Dyrk1a truncation mutants impair neuronal dendritic and spine growth and interfere with postnatal cortical development. Mol. Psychiatry 2018, 23, 747–758. [Google Scholar] [CrossRef] [Green Version]
- Liu, T.; Sims, D.; Baum, B. Parallel RNAi screens across different cell lines identify generic and cell type-specific regulators of actin organization and cell morphology. Genome Biol. 2009, 10, R26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleschevnikov, A.M.; Belichenko, P.V.; Villar, A.J.; Epstein, C.J.; Malenka, R.C.; Mobley, W.C. Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J. Neurosci. 2004, 24, 8153–8160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siarey, R.J.; Carlson, E.J.; Epstein, C.J.; Balbo, A.; Rapoport, S.I.; Galdzicki, Z. Increased synaptic depression in the Ts65Dn mouse, a model for mental retardation in Down syndrome. Neuropharmacology 1999, 38, 1917–1920. [Google Scholar] [CrossRef]
- Siarey, R.J.; Kline-Burgess, A.; Cho, M.; Balbo, A.; Best, T.K.; Harashima, C.; Klann, E.; Galdzicki, Z. Altered signaling pathways underlying abnormal hippocampal synaptic plasticity in the Ts65Dn mouse model of Down syndrome. J. Neurochem. 2006, 98, 1266–1277. [Google Scholar] [CrossRef]
- Martínez-Cué, C.; Martínez, P.; Rueda, N.; Vidal, R.; García, S.; Vidal, V.; Corrales, A.; Montero, J.A.; Pazos, A.; Flórez, J.; et al. Reducing GABAA α5 Receptor-Mediated Inhibition Rescues Functional and Neuromorphological Deficits in a Mouse Model of Down Syndrome. J. Neurosci. 2013, 33, 3953–3966. [Google Scholar] [CrossRef] [Green Version]
- Begenisic, T.; Sansevero, G.; Baroncelli, L.; Cioni, G.; Sale, A. Early environmental therapy rescues brain development in a mouse model of Down syndrome. Neurobiol. Dis. 2015, 82, 409–419. [Google Scholar] [CrossRef]
- Andrade-Talavera, Y.; Benito, I.; Casañas, J.J.; Rodríguez-Moreno, A.; Montesinos, M.L. Rapamycin restores BDNF-LTP and the persistence of long-term memory in a model of Down’s syndrome. Neurobiol. Dis. 2015, 82, 516–525. [Google Scholar] [CrossRef]
- Thomazeau, A.; Lassalle, O.; Iafrati, J.; Souchet, B.; Guedj, F.; Janel, N.; Chavis, P.; Delabar, J.; Manzoni, O.J. Prefrontal deficits in a murine model overexpressing the down syndrome candidate gene dyrk1a. J. Neurosci. 2014, 34, 1138–1147. [Google Scholar] [CrossRef] [Green Version]
- Braudeau, J.; Dauphinot, L.; Duchon, A.; Loistron, A.; Dodd, R.H.; Hérault, Y.; Delatour, B.; Potier, M.C. Chronic Treatment with a Promnesiant GABA-A α5-Selective Inverse Agonist Increases Immediate Early Genes Expression during Memory Processing in Mice and Rectifies Their Expression Levels in a Down Syndrome Mouse Model. Adv. Pharmacol. Sci. 2011, 2011, 153218. [Google Scholar] [CrossRef] [Green Version]
- Duchon, A.; Del Mar Muñiz Moreno, M.; Lorenzo, S.M.; de Souza, M.P.S.; Chevalier, C.; Nalesso, V.; Meziane, H.; de Sousa, P.L.; Noblet, V.; Armspach, J.P.; et al. Multi-influential genetic interactions alter behaviour and cognition through six main biological cascades in Down syndrome mouse models. Hum. Mol. Genet. 2021, 30, 771–788. [Google Scholar] [CrossRef]
- Chen, H.; Firestein, B.L. RhoA regulates dendrite branching in hippocampal neurons by decreasing cypin protein levels. J. Neurosci. 2007, 27, 8378–8386. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, N.; Schohl, A.; Ruthazer, E.S. Neural activity regulates synaptic properties and dendritic structure in vivo through calcineurin/NFAT signaling. Neuron 2009, 62, 655–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, A.; Franciscovich, A.; Bowers, M.; Sandstrom, D.J.; Sanyal, S. NFAT regulates pre-synaptic development and activity-dependent plasticity in Drosophila. Mol. Cell Neurosci. 2011, 46, 535–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisman, J.; Cooper, K.; Sehgal, M.; Silva, A.J. Memory formation depends on both synapse-specific modifications of synaptic strength and cell-specific increases in excitability. Nat. Neurosci. 2018, 21, 309–314. [Google Scholar] [CrossRef] [Green Version]
- Geng, J.; Wang, L.; Lee, J.Y.; Chen, C.K.; Chang, K.T. Phosphorylation of Synaptojanin Differentially Regulates Endocytosis of Functionally Distinct Synaptic Vesicle Pools. J. Neurosci. 2016, 36, 8882–8894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Park, J.; Song, W.J.; Chang, S. Overexpression of Dyrk1A Causes the Defects in Synaptic Vesicle Endocytosis. Neurosignals 2010, 18, 164–172. [Google Scholar] [CrossRef]
- Arque, G.; Casanovas, A.; Dierssen, M. Dyrk1A is dynamically expressed on subsets of motor neurons and in the neuromuscular junction: Possible role in Down syndrome. PLoS ONE 2013, 8, e54285. [Google Scholar] [CrossRef] [Green Version]
- Wiseman, F.K.; Al-Janabi, T.; Hardy, J.; Karmiloff-Smith, A.; Nizetic, D.; Tybulewicz, V.L.; Fisher, E.M.; Strydom, A. A genetic cause of Alzheimer disease: Mechanistic insights from Down syndrome. Nat. Rev. Neurosci. 2015, 16, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Contestabile, A.; Ciani, E.; Contestabile, A. The place of choline acetyltransferase activity measurement in the “Cholinergic hypothesis” of neurodegenerative diseases. Neurochem. Res. 2008, 33, 318–327. [Google Scholar] [CrossRef]
- Granholm, A.C.; Sanders, L.A.; Crnic, L.S. Loss of cholinergic phenotype in basal forebrain coincides with cognitive decline in a mouse model of Down’s syndrome. Exp. Neurol. 2000, 161, 647–663. [Google Scholar] [CrossRef]
- Hunter, C.L.; Isacson, O.; Nelson, M.; Bimonte-Nelson, H.; Seo, H.; Lin, L.; Ford, K.; Kindy, M.S.; Granholm, A.C. Regional alterations in amyloid precursor protein and nerve growth factor across age in a mouse model of Down’s syndrome. Neurosci Res 2003, 45, 437–445. [Google Scholar] [CrossRef]
- Seo, H.; Isacson, O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Exp. Neurol. 2005, 193, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Lockrow, J.; Boger, H.; Gerhardt, G.; Aston-Jones, G.; Bachman, D.; Granholm, A.C. A noradrenergic lesion exacerbates neurodegeneration in a Down syndrome mouse model. J. Alzheimer’s Dis. 2011, 23, 471–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lockrow, J.P.; Fortress, A.M.; Granholm, A.C. Age-related neurodegeneration and memory loss in down syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012, 463909. [Google Scholar] [CrossRef]
- Illouz, T.; Madar, R.; Biragyn, A.; Okun, E. Restoring microglial and astroglial homeostasis using DNA immunization in a Down Syndrome mouse model. Brain Behav. Immun. 2019, 75, 163–180. [Google Scholar] [CrossRef]
- Kelley, C.M.; Powers, B.E.; Velazquez, R.; Ash, J.A.; Ginsberg, S.D.; Strupp, B.J.; Mufson, E.J. Sex differences in the cholinergic basal forebrain in the Ts65Dn mouse model of Down syndrome and Alzheimer’s disease. Brain Pathol. 2014, 24, 33–44. [Google Scholar] [CrossRef] [Green Version]
- Dierssen, M.; Vallina, I.F.; Baamonde, C.; Garcia-Calatayud, S.; Lumbreras, M.A.; Florez, J. Alterations of central noradrenergic transmission in Ts65Dn mouse, a model for Down syndrome. Brain Res. 1997, 749, 238–244. [Google Scholar] [CrossRef]
- Hunter, C.L.; Bimonte-Nelson, H.A.; Nelson, M.; Eckman, C.B.; Granholm, A.C. Behavioral and neurobiological markers of Alzheimer’s disease in Ts65Dn mice: Effects of estrogen. Neurobiol. Aging 2004, 25, 873–884. [Google Scholar] [CrossRef]
- Lomoio, S.; Scherini, E.; Necchi, D. Beta-amyloid overload does not directly correlate with SAPK/JNK activation and tau protein phosphorylation in the cerebellar cortex of Ts65Dn mice. Brain Res. 2009, 1297, 198–206. [Google Scholar] [CrossRef]
- Sansevero, G.; Begenisic, T.; Mainardi, M.; Sale, A. Experience-dependent reduction of soluble β-amyloid oligomers and rescue of cognitive abilities in middle-age Ts65Dn mice, a model of Down syndrome. Exp. Neurol. 2016, 283, 49–56. [Google Scholar] [CrossRef]
- Pop, C.; Timmer, J.; Sperandio, S.; Salvesen, G.S. The apoptosome activates caspase-9 by dimerization. Mol. Cell 2006, 22, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Morrice, N.; Brady, S.; Magee, G.; Pathak, S.; Clarke, P.R. Inhibition of caspase-9 through phosphorylation at Thr 125 by ERK MAPK. Nat. Cell Biol. 2003, 5, 647–654. [Google Scholar] [CrossRef]
- Brady, S.C.; Allan, L.A.; Clarke, P.R. Regulation of caspase 9 through phosphorylation by protein kinase C zeta in response to hyperosmotic stress. Mol. Cell Biol. 2005, 25, 10543–10555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, M.C.; Allan, L.A.; Lickrish, M.; Sampson, C.; Morrice, N.; Clarke, P.R. Protein kinase A regulates caspase-9 activation by Apaf-1 downstream of cytochrome c. J. Biol. Chem. 2005, 280, 15449–15455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Budihardjo, I.; Oliver, H.; Lutter, M.; Luo, X.; Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 1999, 15, 269–290. [Google Scholar] [CrossRef] [Green Version]
- Ferrer, I.; Barrachina, M.; Puig, B.; Martínez de Lagrán, M.; Martí, E.; Avila, J.; Dierssen, M. Constitutive Dyrk1A is abnormally expressed in Alzheimer disease, Down syndrome, Pick disease, and related transgenic models. Neurobiol. Dis. 2005, 20, 392–400. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H.; et al. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegiel, J.; Kaczmarski, W.; Barua, M.; Kuchna, I.; Nowicki, K.; Wang, K.C.; Yang, S.M.; Frackowiak, J.; Mazur-Kolecka, B.; Silverman, W.P.; et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J. Neuropathol. Exp. Neurol. 2011, 70, 36–50. [Google Scholar] [CrossRef] [Green Version]
- Wegiel, J.; Gong, C.X.; Hwang, Y.W. The role of DYRK1A in neurodegenerative diseases. FEBS J. 2011, 278, 236–245. [Google Scholar] [CrossRef]
- Park, J.; Chung, K.C. New Perspectives of Dyrk1A Role in Neurogenesis and Neuropathologic Features of Down Syndrome. Exp. Neurobiol. 2013, 22, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Latour, A.; Gu, Y.; Kassis, N.; Daubigney, F.; Colin, C.; Gausserès, B.; Middendorp, S.; Paul, J.L.; Hindié, V.; Rain, J.C.; et al. LPS-Induced Inflammation Abolishes the Effect of DYRK1A on IkB Stability in the Brain of Mice. Mol. Neurobiol. 2019, 56, 963–975. [Google Scholar] [CrossRef]
- Herault, Y.; Delabar, J.M.; Fisher, E.M.C.; Tybulewicz, V.L.J.; Yu, E.; Brault, V. Rodent models in Down syndrome research: Impact and future opportunities. Dis. Models Mech. 2017, 10, 1165–1186. [Google Scholar] [CrossRef] [Green Version]
- Duchon, A.; Herault, Y. DYRK1A, a Dosage-Sensitive Gene Involved in Neurodevelopmental Disorders, Is a Target for Drug Development in Down Syndrome. Front. Behav. Neurosci. 2016, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Ortiz-Abalia, J.; Sahún, I.; Altafaj, X.; Andreu, N.; Estivill, X.; Dierssen, M.; Fillat, C. Targeting Dyrk1A with AAVshRNA attenuates motor alterations in TgDyrk1A, a mouse model of Down syndrome. Am. J. Hum. Genet. 2008, 83, 479–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Cerro, S.; Rueda, N.; Vidal, V.; Lantigua, S.; Martínez-Cué, C. Normalizing the gene dosage of Dyrk1A in a mouse model of Down syndrome rescues several Alzheimer’s disease phenotypes. Neurobiol. Dis. 2017, 106, 76–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altafaj, X.; Martín, E.D.; Ortiz-Abalia, J.; Valderrama, A.; Lao-Peregrín, C.; Dierssen, M.; Fillat, C. Normalization of Dyrk1A expression by AAV2/1-shDyrk1A attenuates hippocampal-dependent defects in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2013, 52, 117–127. [Google Scholar] [CrossRef] [PubMed]
- De la Torre, R.; De Sola, S.; Pons, M.; Duchon, A.; Martinez de Lagran, M.; Farre, M.; Fito, M.; Benejam, B.; Langohr, K.; Rodriguez, J.; et al. Epigallocatechin-3-gallate, a DYRK1A inhibitor, rescues cognitive deficits in Down syndrome mouse models and in humans. Mol. Nutr. Food Res. 2014, 58, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Guedj, F.; Sébrié, C.; Rivals, I.; Ledru, A.; Paly, E.; Bizot, J.C.; Smith, D.; Rubin, E.; Gillet, B.; Arbones, M.; et al. Green tea polyphenols rescue of brain defects induced by overexpression of DYRK1A. PLoS ONE 2009, 4, e4606. [Google Scholar] [CrossRef] [Green Version]
- Noll, C.; Tlili, A.; Ripoll, C.; Mallet, L.; Paul, J.L.; Delabar, J.M.; Janel, N. Dyrk1a activates antioxidant NQO1 expression through an ERK1/2-Nrf2 dependent mechanism. Mol. Genet. Metab. 2012, 105, 484–488. [Google Scholar] [CrossRef]
- de la Torre, R.; de Sola, S.; Hernandez, G.; Farre, M.; Pujol, J.; Rodriguez, J.; Espadaler, J.M.; Langohr, K.; Cuenca-Royo, A.; Principe, A.; et al. Safety and efficacy of cognitive training plus epigallocatechin-3-gallate in young adults with Down’s syndrome (TESDAD): A double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2016, 15, 801–810. [Google Scholar] [CrossRef]
- Kim, H.; Lee, K.S.; Kim, A.K.; Choi, M.; Choi, K.; Kang, M.; Chi, S.W.; Lee, M.S.; Lee, J.S.; Lee, S.Y.; et al. A chemical with proven clinical safety rescues Down-syndrome-related phenotypes in through DYRK1A inhibition. Dis. Models Mech. 2016, 9, 839–848. [Google Scholar] [CrossRef] [Green Version]
- Neumann, F.; Gourdain, S.; Albac, C.; Dekker, A.D.; Bui, L.C.; Dairou, J.; Schmitz-Afonso, I.; Hue, N.; Rodrigues-Lima, F.; Delabar, J.M.; et al. DYRK1A inhibition and cognitive rescue in a Down syndrome mouse model are induced by new fluoro-DANDY derivatives. Sci. Rep. 2018, 8, 2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedj, F.; Pereira, P.L.; Najas, S.; Barallobre, M.J.; Chabert, C.; Souchet, B.; Sebrie, C.; Verney, C.; Herault, Y.; Arbones, M.; et al. DYRK1A: A master regulatory protein controlling brain growth. Neurobiol. Dis. 2012, 46, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Reeves, R.H.; Irving, N.G.; Moran, T.H.; Wohn, A.; Kitt, C.; Sisodia, S.S.; Schmidt, C.; Bronson, R.T.; Davisson, M.T. A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat. Genet. 1995, 11, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Duchon, A.; Raveau, M.; Chevalier, C.; Nalesso, V.; Sharp, A.J.; Herault, Y. Identification of the translocation breakpoints in the Ts65Dn and Ts1Cje mouse lines: Relevance for modeling Down syndrome. Mamm. Genome 2011, 22, 674–684. [Google Scholar] [CrossRef] [Green Version]
- Mazur-Kolecka, B.; Golabek, A.; Kida, E.; Rabe, A.; Hwang, Y.W.; Adayev, T.; Wegiel, J.; Flory, M.; Kaczmarski, W.; Marchi, E.; et al. Effect of DYRK1A activity inhibition on development of neuronal progenitors isolated from Ts65Dn mice. J. Neurosci. Res. 2012, 90, 999–1010. [Google Scholar] [CrossRef]
- Frost, D.; Meechoovet, B.; Wang, T.; Gately, S.; Giorgetti, M.; Shcherbakova, I.; Dunckley, T. β-carboline compounds, including harmine, inhibit DYRK1A and tau phosphorylation at multiple Alzheimer’s disease-related sites. PLoS ONE 2011, 6, e19264. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Sablin, S.O.; Ramsay, R.R. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch. Biochem. Biophys. 1997, 337, 137–142. [Google Scholar] [CrossRef]
- Drung, B.; Scholz, C.; Barbosa, V.A.; Nazari, A.; Sarragiotto, M.H.; Schmidt, B. Computational & experimental evaluation of the structure/activity relationship of β-carbolines as DYRK1A inhibitors. Bioorgan. Med. Chem. Lett. 2014, 24, 4854–4860. [Google Scholar] [CrossRef]
- Rüben, K.; Wurzlbauer, A.; Walte, A.; Sippl, W.; Bracher, F.; Becker, W. Selectivity Profiling and Biological Activity of Novel β-Carbolines as Potent and Selective DYRK1 Kinase Inhibitors. PLoS ONE 2015, 10, e0132453. [Google Scholar] [CrossRef]
- Pranzatelli, M.R.; Snodgrass, S.R. Harmala alkaloids and related beta-carbolines: A myoclonic model and antimyoclonic drugs. Exp. Neurol. 1987, 96, 703–719. [Google Scholar] [CrossRef]
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, M.; Abeysekera, I.; Thomas, J.; LaCombe, J.; Stancombe, K.; Stewart, R.J.; Dria, K.J.; Wallace, J.M.; Goodlett, C.R.; Roper, R.J. Epigallocatechin-3-gallate (EGCG) consumption in the Ts65Dn model of Down syndrome fails to improve behavioral deficits and is detrimental to skeletal phenotypes. Physiol. Behav. 2017, 177, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Moroy, G.; Paul, J.-L.; Rebillat, A.-S.; Dierssen, M.; de la Torre, R.; Cieuta-Walti, C.; Dairou, J.; Janel, N. Molecular Rescue of Dyrk1A Overexpression Alterations in Mice with Fontup (R) Dietary Supplement: Role of Green Tea Catechins. Int. J. Mol. Sci. 2020, 21, 1404. [Google Scholar] [CrossRef] [Green Version]
- Goodlett, C.R.; Stringer, M.; LaCombe, J.; Patel, R.; Wallace, J.M.; Roper, R.J. Evaluation of the therapeutic potential of Epigallocatechin-3-gallate (EGCG) via oral gavage in young adult Down syndrome mice. Sci. Rep. 2020, 10, 10426. [Google Scholar] [CrossRef]
- Debdab, M.; Carreaux, F.; Renault, S.; Soundararajan, M.; Fedorov, O.; Filippakopoulos, P.; Lozach, O.; Babault, L.; Tahtouh, T.; Baratte, B.; et al. Leucettines, a class of potent inhibitors of cdc2-like kinases and dual specificity, tyrosine phosphorylation regulated kinases derived from the marine sponge leucettamine B: Modulation of alternative pre-RNA splicing. J. Med. Chem. 2011, 54, 4172–4186. [Google Scholar] [CrossRef]
- Li, Z.; Yu, T.; Morishima, M.; Pao, A.; Laduca, J.; Conroy, J.; Nowak, N.; Matsui, S.; Shiraishi, I.; Yu, Y.E. Duplication of the entire 22.9 Mb human chromosome 21 syntenic region on mouse chromosome 16 causes cardiovascular and gastrointestinal abnormalities. Hum. Mol. Genet. 2007, 16, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Naert, G.; Ferré, V.; Meunier, J.; Keller, E.; Malmström, S.; Givalois, L.; Carreaux, F.; Bazureau, J.P.; Maurice, T. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Aβ25-35 peptide administration in mice. Eur. Neuropsychopharmacol. 2015, 25, 2170–2182. [Google Scholar] [CrossRef]
- Gourdain, S.; Dairou, J.; Denhez, C.; Bui, L.C.; Rodrigues-Lima, F.; Janel, N.; Delabar, J.M.; Cariou, K.; Dodd, R.H. Development of DANDYs, new 3,5-diaryl-7-azaindoles demonstrating potent DYRK1A kinase inhibitory activity. J. Med. Chem. 2013, 56, 9569–9585. [Google Scholar] [CrossRef] [PubMed]
- Masaki, S.; Kii, I.; Sumida, Y.; Kato-Sumida, T.; Ogawa, Y.; Ito, N.; Nakamura, M.; Sonamoto, R.; Kataoka, N.; Hosoya, T.; et al. Design and synthesis of a potent inhibitor of class 1 DYRK kinases as a suppressor of adipogenesis. Bioorgan. Med. Chem. 2015, 23, 4434–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonamoto, R.; Kii, I.; Koike, Y.; Sumida, Y.; Kato-Sumida, T.; Okuno, Y.; Hosoya, T.; Hagiwara, M. Identification of a DYRK1A Inhibitor that Induces Degradation of the Target Kinase using Co-chaperone CDC37 fused with Luciferase nanoKAZ. Sci. Rep. 2015, 5, 12728. [Google Scholar] [CrossRef]
- Shen, W.; Tremblay, M.S.; Deshmukh, V.A.; Wang, W.; Filippi, C.M.; Harb, G.; Zhang, Y.Q.; Kamireddy, A.; Baaten, J.E.; Jin, Q.; et al. Small-molecule inducer of β cell proliferation identified by high-throughput screening. J. Am. Chem. Soc. 2013, 135, 1669–1672. [Google Scholar] [CrossRef]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human β-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef]
- Nakano-Kobayashi, A.; Awaya, T.; Kii, I.; Sumida, Y.; Okuno, Y.; Yoshida, S.; Sumida, T.; Inoue, H.; Hosoya, T.; Hagiwara, M. Prenatal neurogenesis induction therapy normalizes brain structure and function in Down syndrome mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10268–10273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souchet, B.; Duchon, A.; Gu, Y.; Dairou, J.; Chevalier, C.; Daubigney, F.; Nalesso, V.; Creau, N.; Yu, Y.; Janel, N.; et al. Prenatal treatment with EGCG enriched green tea extract rescues GAD67 related developmental and cognitive defects in Down syndrome mouse models. Sci. Rep. 2019, 9, 3914. [Google Scholar] [CrossRef] [Green Version]
- Zorrilla de San Martin, J.; Delabar, J.M.; Bacci, A.; Potier, M.C. GABAergic over-inhibition, a promising hypothesis for cognitive deficits in Down syndrome. Free Radic. Biol. Med. 2018, 114, 33–39. [Google Scholar] [CrossRef]
- Contestabile, A.; Magara, S.; Cancedda, L. The GABAergic Hypothesis for Cognitive Disabilities in Down Syndrome. Front. Cell Neurosci. 2017, 11, 54. [Google Scholar] [CrossRef] [Green Version]
- McElyea, S.D.; Starbuck, J.M.; Tumbleson-Brink, D.M.; Harrington, E.; Blazek, J.D.; Ghoneima, A.; Kula, K.; Roper, R.J. Influence of prenatal EGCG treatment and Dyrk1a dosage reduction on craniofacial features associated with Down syndrome. Hum. Mol. Genet. 2016, 25, 4856–4869. [Google Scholar] [CrossRef] [Green Version]
- Stagni, F.; Giacomini, A.; Emili, M.; Trazzi, S.; Guidi, S.; Sassi, M.; Ciani, E.; Rimondini, R.; Bartesaghi, R. Short- and long-term effects of neonatal pharmacotherapy with epigallocatechin-3-gallate on hippocampal development in the Ts65Dn mouse model of Down syndrome. Neuroscience 2016, 333, 277–301. [Google Scholar] [CrossRef]
- Starbuck, J.M.; Llambrich, S.; Gonzàlez, R.; Albaigès, J.; Sarlé, A.; Wouters, J.; González, A.; Sevillano, X.; Sharpe, J.; De La Torre, R.; et al. Green tea extracts containing epigallocatechin-3-gallate modulate facial development in Down syndrome. Sci. Rep. 2021, 11, 4715. [Google Scholar] [CrossRef] [PubMed]
- Xicota, L.; Rodríguez, J.; Langohr, K.; Fitó, M.; Dierssen, M.; de la Torre, R.; TESDAD Study Group. Effect of epigallocatechin gallate on the body composition and lipid profile of down syndrome individuals: Implications for clinical management. Clin. Nutr. 2020, 39, 1292–1300. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Liu, C.; Yu, T.; Zhang, L.; Meng, K.; Xing, Z.; Belichenko, P.V.; Kleschevnikov, A.M.; Pao, A.; Peresie, J.; et al. Genetic dissection of the Down syndrome critical region. Hum. Mol. Genet. 2015, 24, 6540–6551. [Google Scholar] [CrossRef] [PubMed]
- Marechal, D.; Brault, V.; Leon, A.; Martin, D.; Pereira, P.L.; Loaëc, N.; Birling, M.C.; Friocourt, G.; Blondel, M.; Herault, Y. Cbs overdosage is necessary and sufficient to induce cognitive phenotypes in mouse models of Down syndrome and interacts genetically with Dyrk1a. Hum. Mol. Genet. 2019, 28, 1561–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Allele Symbol | Category—Genetic Background | Initial Reference | Repository |
|---|---|---|---|
| Tg(CEPHY152F7)12Hgc | Transgenic YAC Human- FVB or C57BL/6J including TTC3 (tetratricopeptide repeat domain 3), DSCR3 (Down syndrome critical region gene 3), DYRK1A (dual-specificity tyrosine-(Y)-phosphorylation regulated kinase 1A) and the potassium inwardly-rectifying channel, subfamily J, member 6 gene, (KCNJ6) | [57] | INFRAFRONTIER Stock EM:01303 |
| Tg(MT1A-Dyrk1a)9Xest | Transgenic-Heterogenous promoter rat cDNA for Dyrk1a (2 copies) | [58] | Unknown |
| Tg(MT1A-Dyrk1a)33Xest | Transgenic-Heterogenous promoter rat cDNA for Dyrk1a (5 copies) | [58] | Unknown |
| Tg(DYRK1A)36Wjs | Transgenic BAC Human locus for DYRK1A—C57BL/6J coisogenic | [58] | JAX |
| Tg(Dyrk1a)189N3Yah | Transgenic BAC Mouse lous for Dyrk1a- C57BL/6J coisogenic | [59] | INFRAFRONTIER stock EM:02119 |
| Symbol | Name | Cell Function | Ref. |
|---|---|---|---|
| Neurogenesis | |||
| CCND1 | Cyclin D1 | Cell cycle and division (G1 to S phase transition) | [80,82,85] |
| CCNL2 | Cyclin L2 | transcription regulation | [86] |
| CDKN1A | Cyclin Dependent Kinase Inhibitor 1A | Cell cycle and division (G1 to S phase transition) | [30] |
| CDKN1B | Cyclin Dependent Kinase Inhibitor 1B | Cell cycle and division (G1 to S phase transition) | [79,80] |
| LIN52 | Protein lin-52 homolog | Cell cycle, transcription | [83] |
| SNR1 | Integrase interactor | Cell cycle, transcription regulation | [87] |
| FGF2 | Fibroblast Growth Factor 2 | Mitogenic and angiogenic activity | [44] |
| RAS | GTPase Ras | Differentiation, transcription regulation | [88] |
| BRAF | B-Raf Proto-Oncogene, Serine/Threonine Kinase | Differentiation, transcription regulation | [88] |
| MEK1 | Dual specificity mitogen-activated protein kinase 1 | Differentiation, transcription regulation | [88] |
| GLI1 | Glioma-associated oncogene 1 | Differentiation, transcription regulation | [89,90] |
| NOTCH | Notch receptor | Differentiation, transcription regulation | [91] |
| GSK3b | Glycogen synthase kinase 3 β | Differentiation, neurogenesis | [92] |
| FKHR | Forkhead Box O1 | Apoptosis, differentiation, transcription regulation | [93] |
| MAP1B | Microtubule Associated Protein 1B | Microtubule Dynamics, neuritogenesis, axon extension, intracellular transport | [94] |
| RGA4 * | Rho GTPase-activating protein 4 | Actin filaments assembly | [95] |
| CDC42 * | Cell Division Cycle 42 | Actin filaments assembly | [95] |
| N-WASP | Neural Wiskott-Aldrich syndrome protein | Actin filaments assembly, synaptogenesis, cell cycle, cell division, mitosis, transcription | [96] |
| GRB2 | Growth Factor Receptor Bound Protein 2 | Cell differentiation | [97,98] |
| Adult brain function/ Synaptic activity | |||
| YWHAZ | 14-3-3 kinase-binding protein | Signal transduction, spine maturation | [40,99] |
| NFATC | Nuclear factor of activated T cells | Synaptic plasticity, transcription regulation | [100,101,102] |
| RCAN1 | Regulator of calcineurin 1 | calcineurin-NFAT signaling cascade | [103,104] |
| CREB | CAMP-Responsive Element-Binding Protein | Synaptic plasticity, differentiation, transcription regulation | [44] |
| RNAPII | RNA polymerase II | Transcription | [105] |
| DNM1 | Dynamin 1 | Clathrin-mediated vesicle endocytosis | [106,107,108] |
| AMPH | Amphiphysin 1 | Clathrin-mediated vesicle endocytosis | [66,107] |
| SH3GL2 | SH3-domain GRB2-like 2/ Endophilin 1 | Clathrin-mediated vesicle endocytosis | [109] |
| SYNJ1 | Synaptojanin 1 | Clathrin-mediated vesicle endocytosis | [107,110,111] |
| SNAP91 | Clathrin Coat Assembly Protein AP180 | Clathrin-mediated vesicle endocytosis | [107] |
| GSK3β | Glycogensynthasekinase3 β | Synaptic plasticity | [112] |
| MAP1A | Microtubule Associated Protein 1A | Clathrin-mediated vesicle endocytosis | [107] |
| MAP1B | Microtubule Associated Protein 1B | Axon extension, intracellular transport | [94] |
| MAP2 | Microtubule Associated Protein 2 | Clathrin-mediated vesicle endocytosis | [107] |
| TUBB | Tubulin β Class I/β-tubulin | Microtubule | [113] |
| AP2A1 | Adaptor Related Protein Complex 2 Subunit α/1α-adaptin | Clathrin-mediated vesicle endocytosis | [107] |
| AP1B1 | Adaptor Related Protein Complex 2 Subunit β 1/β-adaptin | Clathrin-mediated vesicle endocytosis | [107] |
| STXBP1 | Syntaxin Binding Protein 1 | SNARE-mediated endocytosis | [114] |
| SYN1 | Synapsin I | Neurotransmitter release | [52] |
| GRIN2A | Glutamate receptor, ionotropic, NMDA2A | Receptor, ion transport | [115,116] |
| GRIN2B | Glutamate receptor, ionotropic, NMDA2B | Receptor, ion transport | [117] |
| SPRY2 | Sprouty 2 | Receptor trafficking | [51] |
| SPRED1 | Sprouty Related EVH1 Domain Containing 1 | RTK/Ras/ERK pathway | [43] |
| SPRED2 | Sprouty Related EVH1 Domain Containing 2 | RTK/Ras/ERK pathway | [43] |
| CAMK2 | Calcium/Calmodulin Dependent Protein Kinase II | Synaptic plasticity | [118] |
| DLG4 | discs large MAGUK scaffold protein 4 /Post-Synaptic Density Protein 95 | Synaptic plasticity | [117] |
| SYNGAP1 | Synaptic Ras GTPase Activating Protein 1 | Synaptic plasticity | [117] |
| SWI/SNF | SWI/SNF-Related Matrix-Associated Actin-Dependent Regulator Of Chromatin Subfamily | Chromatin remodeling | [119] |
| ARIP4 | Androgen Receptor-Interacting Protein 4 | Chromatin remodeling, ATP, DNA and Nucleotide-binding | [61] |
| H3 | Clustered Histone 14 | Chromatin, chromosomal stability | [120] |
| SF3B1 | Splicing factor 3b, subunit 1 | mRNA processing, mRNA splicing | [68] |
| SRSF1 | Serine/Arginine Rich Splicing Factor 1 | mRNA processing, splicing and transport | [121,122,123,124] |
| SRSF2 | Serine/arginine-rich splicing factor 2 | mRNA processing, splicing and transport | [121,122,123,124] |
| SRSF6 | Serine/arginine-rich splicing factor 6 | mRNA processing, splicing and transport | [121,122,123,124] |
| SRSF7 | Serine/arginine-rich splicing factor 6 | mRNA processing, splicing and transport | [121,122,123,124] |
| RNF169 | Ring Finger Protein 169 | Repair following DNA damage | [125,126,127] |
| Neurodegeneration/Cell death | |||
| MAPT | Microtubule-associated protein | Microtubule dynamics | [64,128,129] |
| APP | Amyloid precursor protein | Apoptosis, cell adhesion, endocytosis | [130,131] |
| CASP9 | Caspase 9 | Apoptosis | [132,133] |
| SNCA | α-synuclein | SNARE-mediated endocytosis | [134] |
| LATS2 | Large Tumor Suppressor Kinase 2 | Cell proliferation, apoptosis | [45] |
| SIRT1 | Sirtuin 1 | Histone deacetylation/ cell survival | [135] |
| SEPT4 | Septin 4 | Nucleotide-binding, | [136] |
| PSEN1 | Presenilin 1 | Neuronal degeneration, apoptosis, cell adhesion, Notch signaling | [137] |
| NEP1 | Neprilysin | Aβ-degrading enzyme | [138,139,140] |
| IDE | Insulin Degrading Enzyme | Intercellular peptide signaling | [138,139,140] |
| PARK2 | Parkin | Autophagy, transcription regulation | [141] |
| HIP1 | Huntingtin interacting protein 1 | Apoptosis, differentiation, endocytosis, transcription regulation | [142] |
| HAP1 | Huntingtin Associated Protein 1 | Vesicular trafficking | [143] |
| DCAF7 | DDB1 and CUL4 associated factor 7 | Ubl conjugation pathway | [53,54,143] |
| CAPN1 | Calpain 1 | Protease activity | [47] |
| STAT3A | Signal Transducer And Activator Of Transcription | Activator neuroinflammation | [48] |
| PHYHIP | Phytanoyl-CoA α-hydroxylase associated protein 1 | DYRK1A interacting protein | [55] |
| DYRK1A Inhibitors | Animal Models Tested | Effects on Animal Models | Clinical Trial | Ref. |
|---|---|---|---|---|
| Harmine | Tg(Dyrk1a)189N3Yah | Decrease in homocysteine and ERK ½ liver | No information | [199] |
| 2 months age, 10 mg/kg, intra-periotoneal (i. p.) daily injection | ||||
| EGCG | YACtg152F7 | Restoration of brain weight and morphology and object recognition memory | [198] | |
| From gestation to adulthood, orally administratation at 0.6–1 mg/day or 1.2 mg/day | ||||
| Tg(Dyrk1a)189N3Yah | Normalization of the long term potentiation and spine density in the prefrontal cortex. | [158] | ||
| 3–4 months age, oral administration at 200 mg/kg/day during 4–6 weeks. | ||||
| Tg(Dyrk1a)189N3Yah | Improvement of novel object recognition memory and spatial learning and memory. | 2014, 12 months, Barcelona, Spain, Phase 2 trial. done on 84 people aged between 16 and 34 years old which received a dose of 9 mg/kg/day of EGCG. Improvement of visual recognition memory, ability to control inhibition and their adaptative behavior. | [197] | |
| 3 months age, administration in drinking water during 1 month | ||||
| Ts(1716)65Dn | Spatial learning and memory recovery. | |||
| 3 months age, administration in drinking water during 1 month | ||||
| 2016, Barcelona, Spain, Phase 2 trial. | [200] | |||
| Realize on 76 children (6 and 12 years old) which recerved a daily dose of 10 mg/kg/day of EGCG | ||||
| Leucettine 41 | Tg(Dyrk1a)189N3Yah | Improvement of novel object recognition memory | Not done | [52] |
| 3 months age, i. p., administration during 19 days at 20 mg/kg | ||||
| Ts(1716)65Dn | Improvement of novel object recognition memory | |||
| 3 months age, i. p., administration during 19 days at 20 mg/kg | ||||
| Dp(16)1Yey | Improvement of novel object recognition memory | |||
| 3 months age, i. p., administration during 19 days at 20 mg/kg | Change in functional connectivity in brain area | |||
| CX-4945 | Tg(DYRK1A)36Wjs | Normalization of TAU-pThr212 | Not tested | [201] |
| 8 to 10 weeks old, Acute oral administration of 75 mg/kg | ||||
| Drosophilia larvae | Rescue eyes and wings deformities | |||
| Administration of 100 nM from embryo stage until adulthood stage | ||||
| F-DANDY | Ts(1716)65Dn | Rescue of spatial learning deficit | Not tested | [202] |
| Administration by i. p., at 20 mg/kg/day | ||||
| INDY | Xenopus laevis | Eyes deformities correction | Not tested | [9] |
| Embryos treated with 2.5 µM | ||||
| FINDY | Xenopus | Restoration of neural tissu malformation | Not tested | [39] |
| Embryos treated with 2.5 µM |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atas-Ozcan, H.; Brault, V.; Duchon, A.; Herault, Y. Dyrk1a from Gene Function in Development and Physiology to Dosage Correction across Life Span in Down Syndrome. Genes 2021, 12, 1833. https://doi.org/10.3390/genes12111833
Atas-Ozcan H, Brault V, Duchon A, Herault Y. Dyrk1a from Gene Function in Development and Physiology to Dosage Correction across Life Span in Down Syndrome. Genes. 2021; 12(11):1833. https://doi.org/10.3390/genes12111833
Chicago/Turabian StyleAtas-Ozcan, Helin, Véronique Brault, Arnaud Duchon, and Yann Herault. 2021. "Dyrk1a from Gene Function in Development and Physiology to Dosage Correction across Life Span in Down Syndrome" Genes 12, no. 11: 1833. https://doi.org/10.3390/genes12111833
APA StyleAtas-Ozcan, H., Brault, V., Duchon, A., & Herault, Y. (2021). Dyrk1a from Gene Function in Development and Physiology to Dosage Correction across Life Span in Down Syndrome. Genes, 12(11), 1833. https://doi.org/10.3390/genes12111833