
Chromosome Genome Assembly of Cromileptes altivelis Reveals Loss of Genome Fragment in Cromileptes Compared with Epinephelus Species


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, Library Construction and Sequencing
2.2. Genome Assembly
2.3. Pseudochromosome Construction
2.4. Repeat Annotation
2.5. Genome Prediction and Annotation
2.6. Evolution Analyses
3. Results
3.1. Sequencing and Genome Assembly
3.2. Annotation
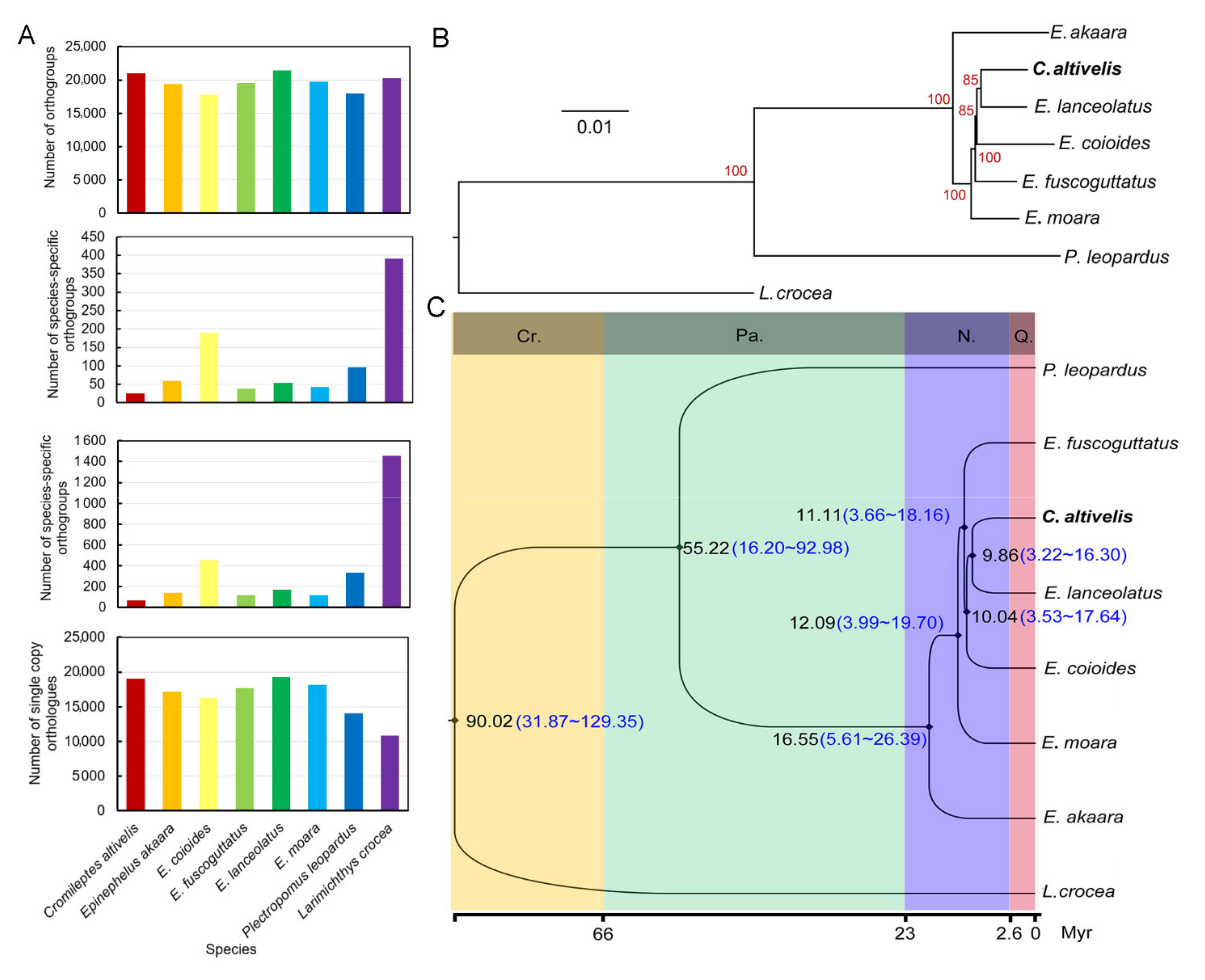
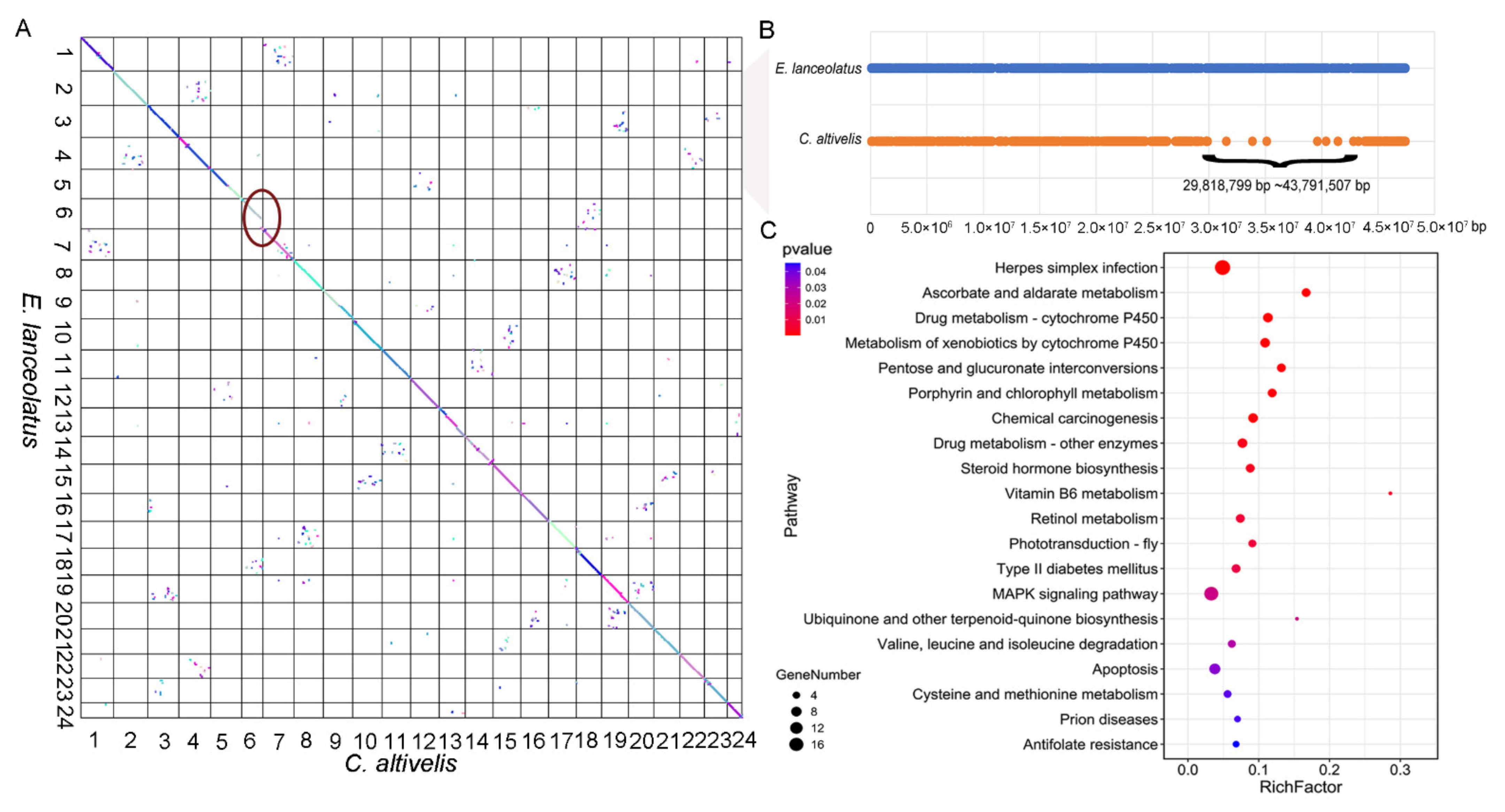
3.3. Evolution analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Froese, R.; Pauly, D. FishBase. World Wide Web Electronic Publication; Version (12/2019). 2019. Available online: www.fishbase.org (accessed on 20 March 2021).
- Shapawi, R.; Mustafa, S.; Ng, W.K. Effects of dietary carbohydrate source and level on growth, feed utilization, and body composition of the humpback grouper, Cromileptes altivelis (Valenciennes). J. Appl. Aquac. 2011, 23, 112–121. [Google Scholar] [CrossRef]
- Zhuang, X.; Qu, M.; Zhang, X.; Ding, S. A Comprehensive Description and Evolutionary Analysis of 22 Grouper (Perciformes, Epinephelidae) Mitochondrial Genomes with Emphasis on Two Novel Genome Organizations. PLoS ONE 2013, 8, e73561. [Google Scholar] [CrossRef] [Green Version]
- Ding, S.; Zhuang, X.; Guo, F.; Wang, J.; Su, Y.; Zhang, Q.; Li, Q. Molecular phylogenetic relationships of China Seas groupers based on cytochrome b gene fragment sequences. Sci. China Ser. C Life Sci. 2006, 49, 235–242. [Google Scholar] [CrossRef]
- Ma, K.Y.; Craig, M.T.; Choat, J.H.; van Herwerden, L. The historical biogeography of groupers: Clade diversification patterns and processes. Mol. Phylogenet. Evol. 2016, 100, 21–30. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, T.; Chen, J.; Wu, L.; Wu, X.; Zhang, W.; Luo, J.; Xia, J.; Meng, Z.; Liu, X. Whole-genome sequencing of brown-marbled grouper (Epinephelus fuscoguttatus) provides insights into adaptive evolution and growth differences. Mol. Ecol. Resour. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ge, H.; Lin, K.; Shen, M.; Wu, S.; Wang, Y.; Zhang, Z.; Wang, Z.; Zhang, Y.; Huang, Z.; Zhou, C.; et al. De novo assembly of a chromosome-level reference genome of red-spotted grouper (Epinephelus akaara) using nanopore sequencing and Hi-C. Mol. Ecol. Resour. 2019, 19, 1461–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Q.; Gao, H.; Zhang, Y.; Fan, G.; Xu, H.; Zhai, J.; Xu, W.; Chen, Z.; Zhang, H.; Liu, S.; et al. A chromosome-level genome assembly of the giant grouper (Epinephelus lanceolatus) provides insights into its innate immunity and rapid growth. Mol. Ecol. Resour. 2019, 19, 1322–1332. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wu, L.N.; Chen, J.F.; Wu, X.; Xia, J.H.; Meng, Z.N.; Liu, X.C.; Lin, H.R. Whole-genome sequencing of leopard coral grouper (Plectropomus leopardus) and exploration of regulation mechanism of skin color and adaptive evolution. Zool. Res. 2020, 41, 328–340. [Google Scholar] [CrossRef]
- Zhou, Q.; Gao, H.; Xu, H.; Lin, H.; Chen, S. A Chromosomal-scale Reference Genome of the Kelp Grouper Epinephelus moara. Mar. Biotechnol. 2021, 23, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shi, Y.; Yuan, J.; Galaxy, Y.; Zhang, H.; Li, N.; Li, Z.; Chen, Y.; Mu, D.; Parkin, I. Estimation of genomic characteristics by analyzing k-mer frequency in de novo genome projects. arXiv 2013, arXiv:1308.2012. [Google Scholar]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive κ-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, M.; Baldwin-Brown, J.G.; Long, A.D.; Emerson, J.J. Contiguous and accurate de novo assembly of metazoan genomes with modest long read coverage. Nucleic Acids Res. 2016, 44, e147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Burton, J.N.; Adey, A.; Patwardhan, R.P.; Qiu, R.; Kitzman, J.O.; Shendure, J. Chromosome-scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 2013, 31, 1119–1125. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Su, Y.; Liu, L.; Yang, J.; Zhu, Y.; Li, C. Structure and evolution of full-length LTR retrotransposons in rice genome. Plant Syst. Evol. 2010, 287, 19–28. [Google Scholar] [CrossRef]
- Edgar, R.C.; Myers, E.W. PILER: Identification and classification of genomic repeats. Bioinformatics 2005, 21, i152–i158. [Google Scholar] [CrossRef]
- Hoede, C.; Arnoux, S.; Moisset, M.; Chaumier, T.; Inizan, O.; Jamilloux, V.; Quesneville, H. PASTEC: An automatic transposable element classification tool. PLoS ONE 2014, 9, e91929. [Google Scholar] [CrossRef] [Green Version]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 11–14. [Google Scholar] [CrossRef]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- Lowe, T.M.; Eddy, S.R. TRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1996, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Stærfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Burge, C.; Karlin, S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: A b initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S.L. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Alioto, T.; Blanco, E.; Parra, G.; Guigó, R. Using geneid to Identify Genes. Curr. Protoc. Bioinform. 2018, 64. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [Green Version]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pimentel, H.; Salzberg, S.L.; Rinn, J.L.; Pachter, L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.A.; Haas, B.J.; Hamilton, J.P.; Mount, S.M.; Robin, C.R. Comprehensive analysis of alternative splicing in rice and comparative analyses with Arabidopsis. BMC Genom. 2006, 7, 327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, S.; Lomsadze, A.; Borodovsky, M. Identification of protein coding regions in RNA transcripts. Nucleic Acids Res. 2015, 43, e78. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Robin, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [Green Version]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.L.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimmer, E.C.; Huntley, R.P.; Alam-Faruque, Y.; Sawford, T.; O’Donovan, C.; Martin, M.J.; Bely, B.; Browne, P.; Chan, W.M.; Eberhardt, R.; et al. The UniProt-GO Annotation database in 2011. Nucleic Acids Res. 2012, 40, D565–D570. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Boeckmann, B.; Bairoch, A.; Apweiler, R.; Blatter, M.C.; Estreicher, A.; Gasteiger, E.; Martin, M.J.; Michoud, K.; O’Donovan, C.; Phan, I.; et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003, 31, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Rozewicki, J.; Li, S.; Amada, K.M.; Standley, D.M.; Katoh, K. MAFFT-DASH: Integrated protein sequence and structural alignment. Nucleic Acids Res. 2019, 47, W5–W10. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z. Paml: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Suleski, M.; Hedges, S.B. TimeTree: A Resource for Timelines, Timetrees, and Divergence Times. Mol. Biol. Evol. 2017, 34, 1812–1819. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Saltzman, A.; Stone, M.; Franks, C.; Searfoss, G.; Munro, R.; Jaye, M.; Ivashchenko, Y. Cloning and characterization of human Jak-2 kinase: High mRNA expression in immune cells and muscle tissue. Biochem. Biophys. Res. Commun. 1998, 246, 627–633. [Google Scholar] [CrossRef]
- Jäkel, H.; Weinl, C.; Hengst, L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene 2011, 30, 3502–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berry, D.C.; Jin, H.; Majumdar, A.; Noy, N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 2011, 108, 4340–4345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [Green Version]
- Alonen, A.; Finel, M.; Kostiainen, R. The human UDP-glucuronosyltransferase UGT1A3 is highly selective towards N2 in the tetrazole ring of losartan, candesartan, and zolarsartan. Biochem. Pharmacol. 2008, 76, 763–772. [Google Scholar] [CrossRef]
- Miley, M.J.; Zielinska, A.K.; Keenan, J.E.; Bratton, S.M.; Radominska-Pandya, A.; Redinbo, M.R. Crystal structure of the cofactor-binding domain of the human phase II drug-metabolism enzyme UDP-glucuronosyltransferase 2B7. J. Mol. Biol. 2007, 369, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Perreault, M.; Gauthier-Landry, L.; Trottier, J.; Verreault, M.; Caron, P.; Finel, M.; Barbier, O. The Human UDP-glucuronosyltransferase UGT2A1 and UGT2A2 enzymes are highly active in bile acid glucuronidation. Drug Metab. Dispos. 2013, 41, 1616–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, J.D.; Pulford, D.J. The glut athione s-transferase supergene family: Regulation of GST and the contribution of the lsoenzymes to cancer chemoprotection and drug resistance part I. Crit. Rev. Biochem. Mol. Biol. 1995, 30, 445–520. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Raw Data | Reads Number | Reads Base (bp) | N50 (bp) | Max Length (bp) | GC Content (%) |
|---|---|---|---|---|---|
| Illumina data for annotation | 39,031,897 | 11,658,953,034 | 150 | 150 | 49.7 |
| Illumina data for survey | 1,307,931,088 | 196,189,663,200 | 150 | 150 | 41.2 |
| PacBio data | 6,672,321 | 119,331,383,944 | 27,957 | 224,636 | 41.0 |
| HiC data | 336,775,366 | 100,882,400,830 | 150 | 150 | 42.6 |
| Assembled data | Contig or Scaffold number | Genome size (bp) | |||
| Survey | - | ~1,070,000,000 | - | - | 41.2 |
| Contig assembled using PacBio | 470 | 1,044,397,337 | 18,092,086 | 49,150,803 | 41.3 |
| Contig assembled using PacBio+Hi-C | 283 | 1,013,358,489 | 18,269,829 | 49,150,803 | 41.2 |
| Scaffold | 164 | 1,013,370,389 | 52,436,080 | 41.2 | |
| Chromosome | 24 | 1,010,598,072 | 43,466,351 | 52,436,080 | 41.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Y.; Wu, L.; Weng, Z.; Wu, X.; Wang, X.; Xia, J.; Meng, Z.; Liu, X. Chromosome Genome Assembly of Cromileptes altivelis Reveals Loss of Genome Fragment in Cromileptes Compared with Epinephelus Species. Genes 2021, 12, 1873. https://doi.org/10.3390/genes12121873
Yang Y, Wu L, Weng Z, Wu X, Wang X, Xia J, Meng Z, Liu X. Chromosome Genome Assembly of Cromileptes altivelis Reveals Loss of Genome Fragment in Cromileptes Compared with Epinephelus Species. Genes. 2021; 12(12):1873. https://doi.org/10.3390/genes12121873
Chicago/Turabian StyleYang, Yang, Lina Wu, Zhuoying Weng, Xi Wu, Xi Wang, Junhong Xia, Zining Meng, and Xiaochun Liu. 2021. "Chromosome Genome Assembly of Cromileptes altivelis Reveals Loss of Genome Fragment in Cromileptes Compared with Epinephelus Species" Genes 12, no. 12: 1873. https://doi.org/10.3390/genes12121873
APA StyleYang, Y., Wu, L., Weng, Z., Wu, X., Wang, X., Xia, J., Meng, Z., & Liu, X. (2021). Chromosome Genome Assembly of Cromileptes altivelis Reveals Loss of Genome Fragment in Cromileptes Compared with Epinephelus Species. Genes, 12(12), 1873. https://doi.org/10.3390/genes12121873