
Co-Occurrence of Fragile X Syndrome with a Second Genetic Condition: Three Independent Cases of Double Diagnosis

 ,
, 

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Molecular Diagnosis
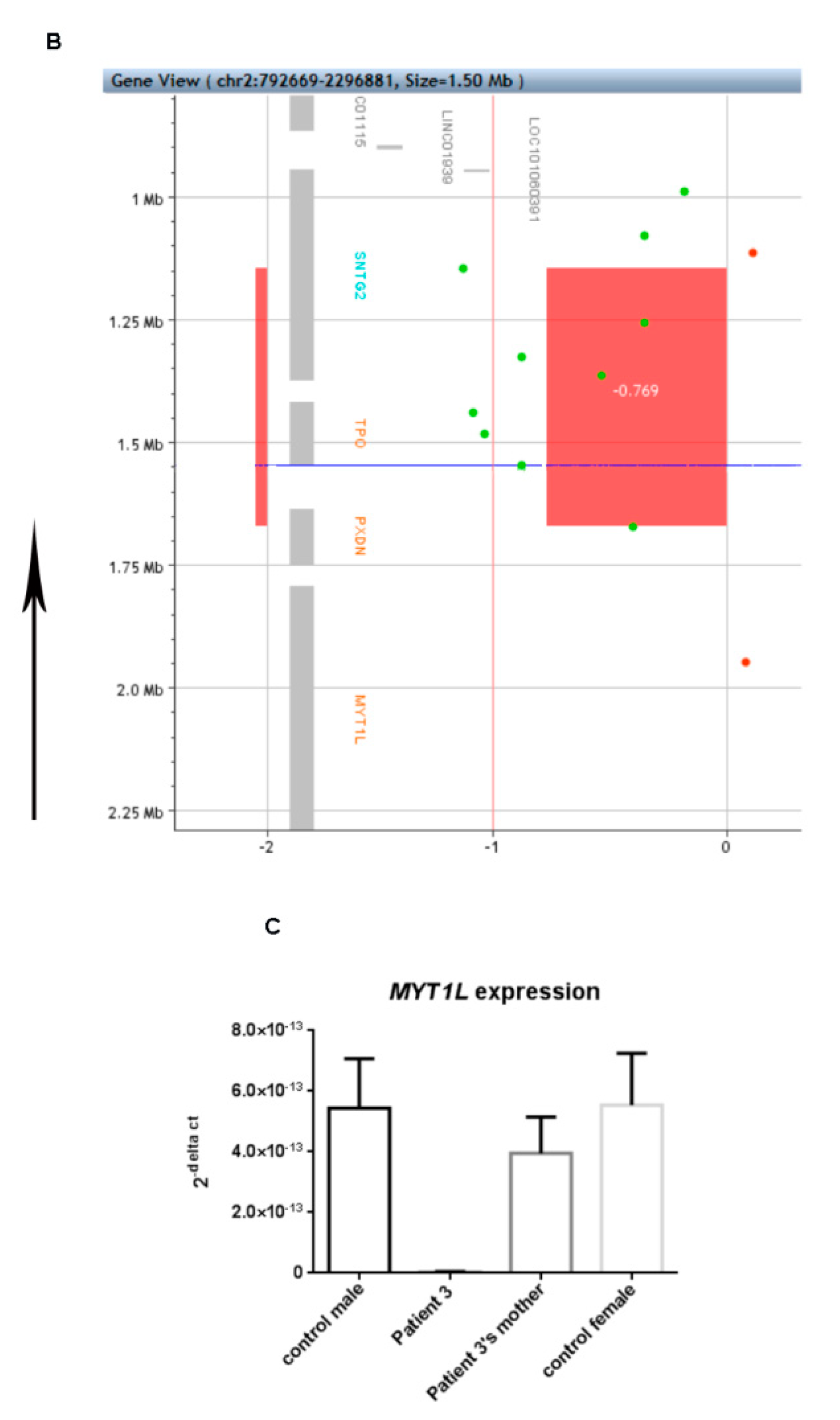
2.3. Expression Analysis of MYT1L
3. Clinical Reports
3.1. Patient 1
3.2. Patient 2
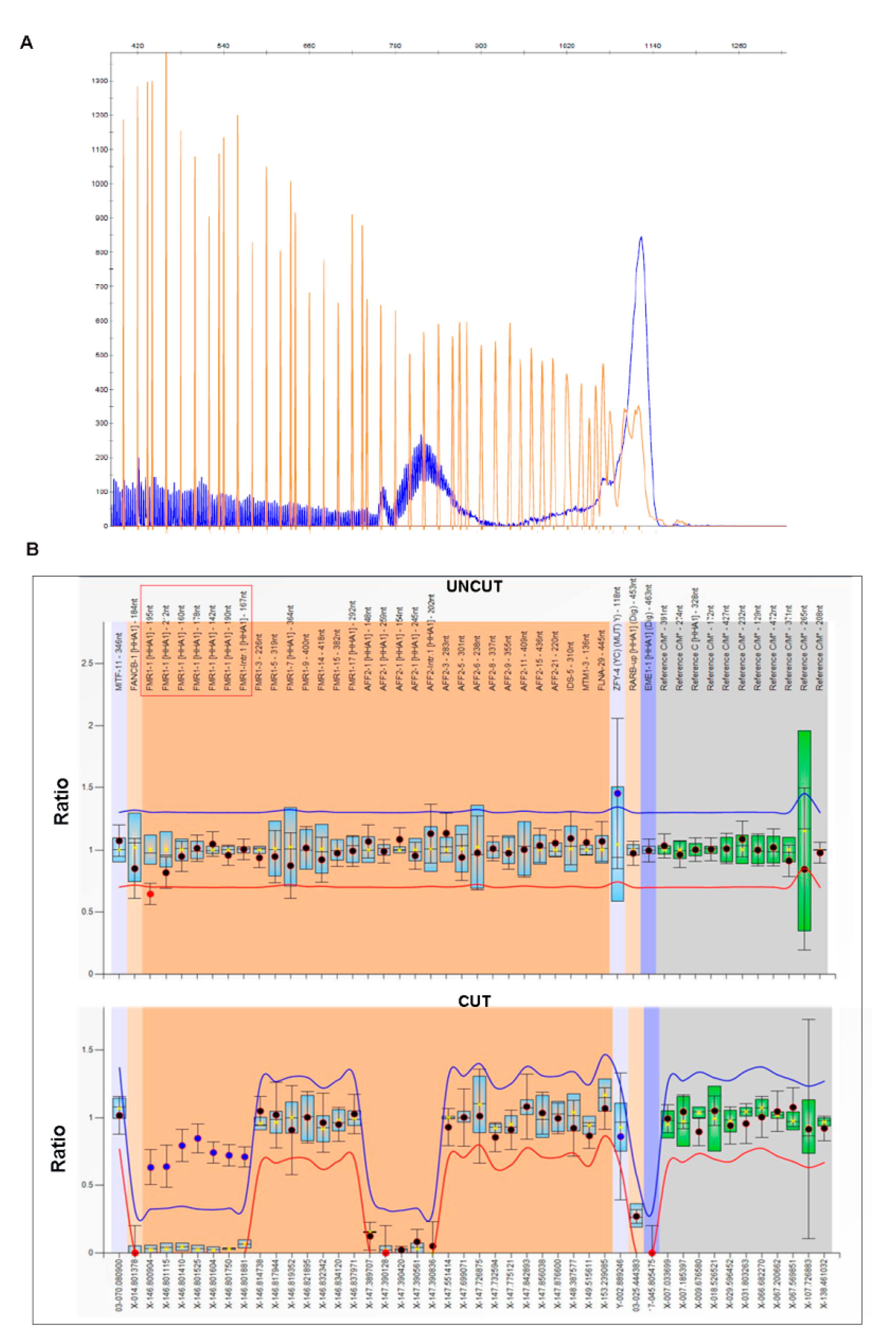
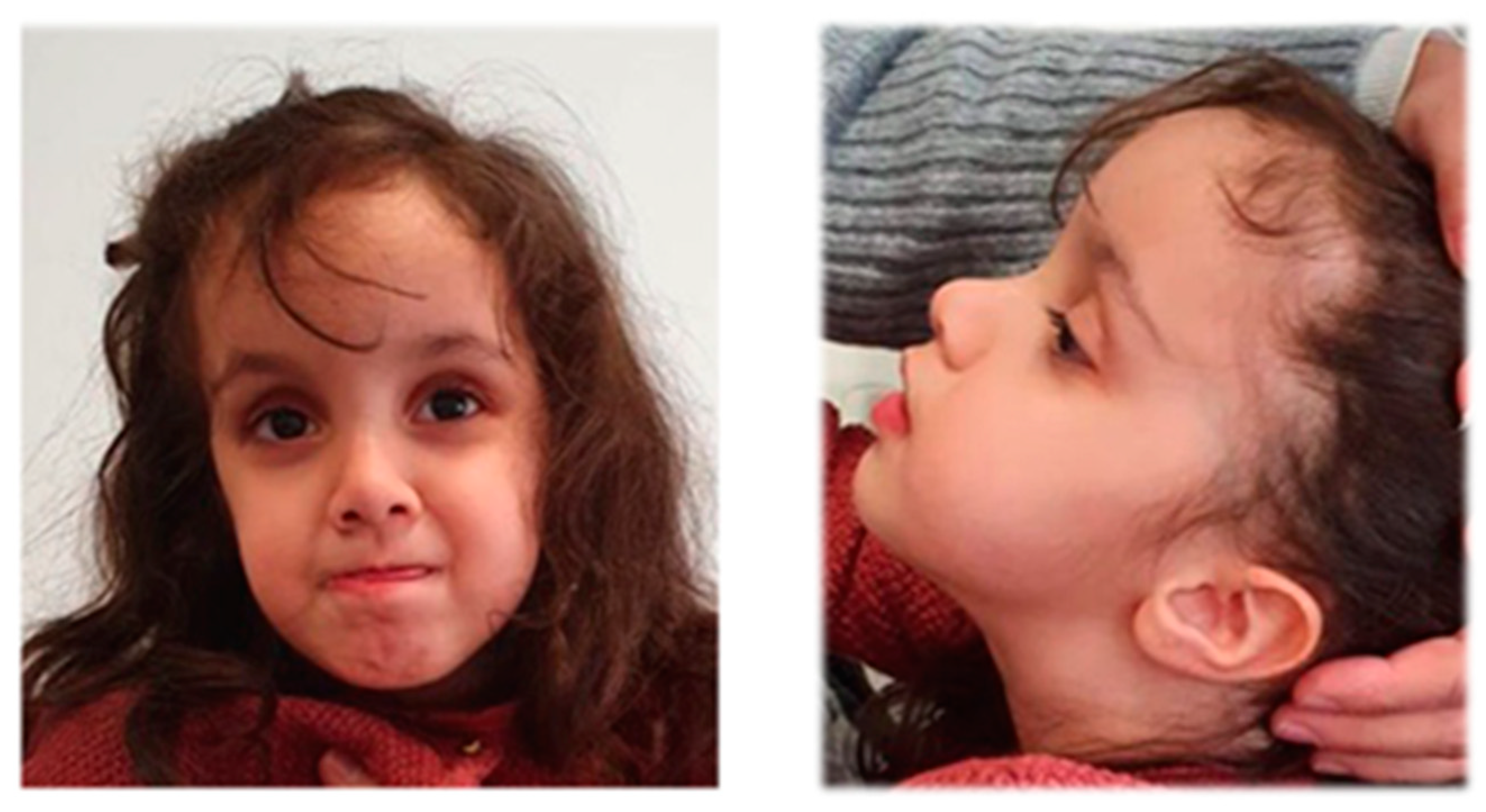
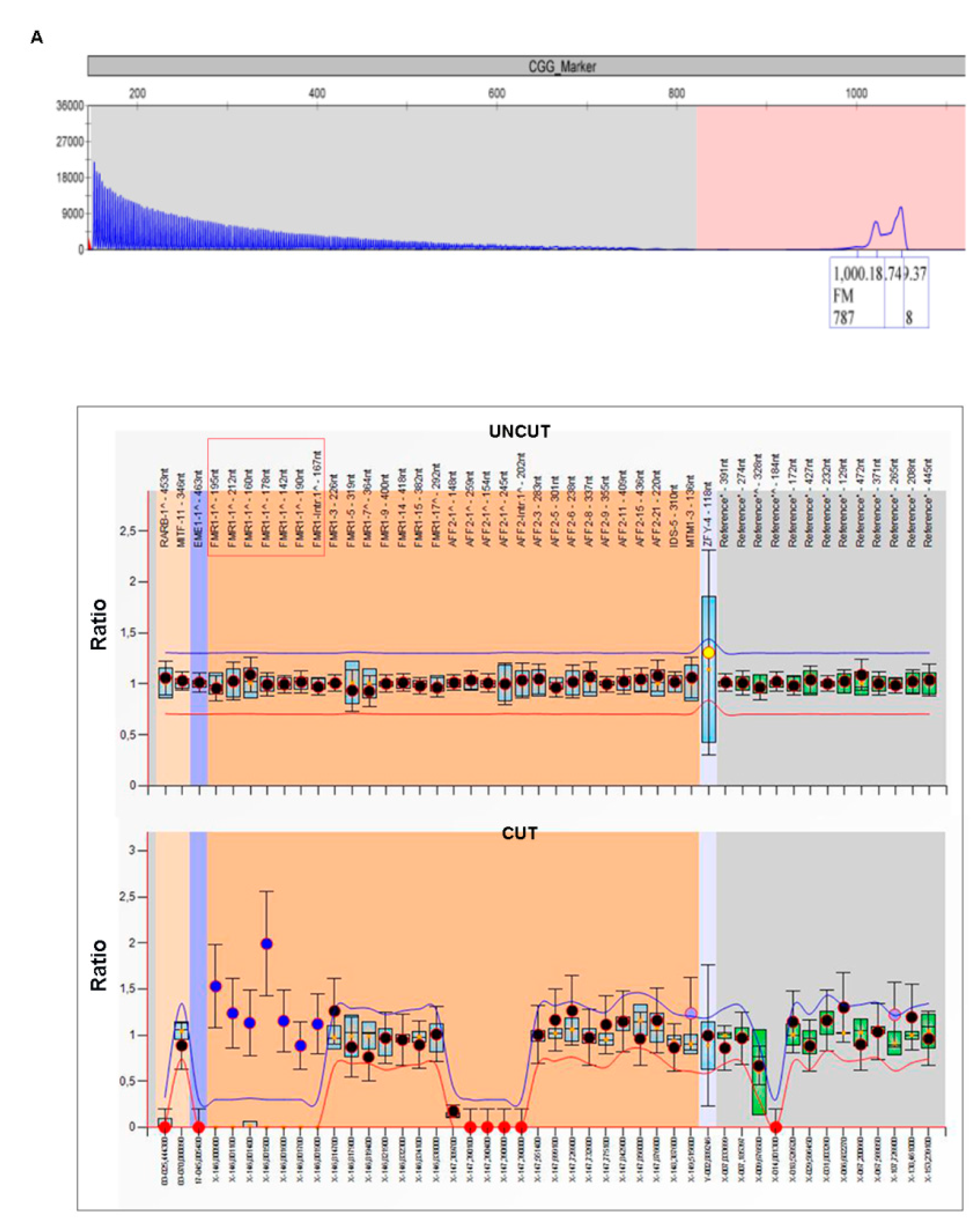
3.3. Patient 3
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunter, J.; Rivero-Arias, O.; Angelov, A.; Kim, E.; Fotheringham, I.; Leal, J. Epidemiology of fragile X syndrome: A systematic review and meta-analysis. Am. J. Med. Genet. Part A 2014, 164, 1648–1658. [Google Scholar] [CrossRef]
- Coffee, B.; Keith, K.; Albizua, I.; Malone, T.; Mowrey, J.; Sherman, S.L.; Warren, S.T. Incidence of fragile X syndrome by newborn screening for methylated FMR1 DNA. Am. J. Hum. Genet. 2009, 85, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Pirozzi, F.; Tabolacci, E.; Neri, G. The FRAXopathies: Definition, overview, and update. Am. J. Med. Genet. Part A 2011, 155, 1803–1816. [Google Scholar] [CrossRef] [PubMed]
- Lahbib, S.; Trabelsi, M.; Dallali, H.; Sakka, R.; Bourourou, R.; Kefi, R.; Mrad, R.; Abdelhak, S.; Gaddour, N. Novel MED12 variant in a multiplex Fragile X syndrome family: Dual molecular etiology of two X-linked intellectual disabilities with autism in the same family. Mol. Biol. Rep. 2019, 46, 4185–4193. [Google Scholar] [CrossRef] [PubMed]
- Saldarriaga, W.; Ruiz, F.A.; Tassone, F.; Hagerman, R. Down Syndrome and Fragile X Syndrome in a Colombian Woman: Case Report. J. Appl. Res. Intellect. Disabil. 2017, 30, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Dobkin, C.; Radu, G.; Ding, X.H.; Brown, W.T.; Nolin, S.L. Fragile X prenatal analyses show full mutation females at high risk for mosaic Turner syndrome: Fragile X leads to chromosome loss. Am. J. Med. Genet. Part A 2009, 149, 2152–2157. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Kidd, S.A.; Andrews, H.F.; Budimirovic, D.B.; Esler, A.; Haas-Givler, B.; Stackhouse, T.; Riley, C.; Peacock, G.; Sherman, S.L.; et al. Autism Spectrum Disorder in Fragile X Syndrome: Cooccurring Conditions and Current Treatment. Pediatrics 2017, 139, S194–S206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.Y.; Kuban, K.C.K.; Allred, E.; Shapiro, F.; Darras, B.T. Association of Duchenne Muscular Dystrophy with Autism Spectrum Disorder. J. Child. Neurol. 2005, 20, 790–795. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef]
- Emery, A.E. The muscular dystrophies. Lancet 2002, 359, 687–695. [Google Scholar] [CrossRef]
- Naidoo, M.; Anthony, K. Dystrophin Dp71 and the neuropathophysiology of Duchenne Muscular Dystrophy. Mol. Neurobiol. 2020, 57, 1748–1767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.-H.; Fang, P.; Adesina, A.M.; Furman, P.; Johnston, J.J.; Biesecker, L.G.; Brown, C.W. Molecular characterization of co-occurring Duchenne muscular dystrophy and X-linked Oculo-Facio-Cardio-Dental syndrome in a girl. Am. J. Med. Genet. Part A 2009, 149A, 1249–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katayama, Y.; Tran, V.K.; Hoan, N.T.; Zhang, Z.; Goji, K.; Yagi, M.; Takeshima, Y.; Saiki, K.; Nhan, N.T.; Matsuo, M. Co-occurrence of Mutations in Both Dystrophin and Androgen-Receptor Genes Is a Novel Cause of Female Duchenne Muscular Dystrophy. Hum. Genet. 2006, 119, 516–519. [Google Scholar] [CrossRef] [PubMed]
- Natori, N. A Case of Atypical Duchenne Type Muscular Dystrophy with Fragile X. Jpn. J. Hum. Genet. 1992, 37, 235–239. [Google Scholar] [CrossRef]
- Todorova, A.; Litvinenko, I.; Todorov, T.; Tincheva, R.; Avdjieva, D.; Tincheva, S.; Mitev, V. A Family with Fragile X Syndrome, Duchenne Muscular Dystrophy and Ichthyosis Transmitted by an Asymptomatic Carrier. Clin. Genet. 2014, 85, 286–289. [Google Scholar] [CrossRef]
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Houge, G.; Haesen, D.; Vissers, L.E.L.M.; Mehta, S.; Parker, M.J.; Wright, M.; Vogt, J.; McKee, S.; Tolmie, J.L.; Cordeiro, N.; et al. B56-delta-related protein phosphatase 2A dysfunction identified in patients with intellectual disability. J. Clin. Investig. 2015, 125, 3051–3062. [Google Scholar] [CrossRef] [Green Version]
- Loveday, C.; Tatton-Brown, K.; Clarke, M.; Westwood, I.; Renwick, A.; Ramsay, E.; Nemeth, A.; Campbell, J.; Joss, S.; Gardner, M.; et al. Mutations in the PP2A regulatory subunit B family genes PPP2R5B, PPP2R5C and PPP2R5D cause human overgrowth. Hum. Molec. Genet. 2015, 24, 4775–4779. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.Y.; Wirth, T.; Hubsch, C.; Németh, A.H.; Okur, V.; Anheim, M.; Drouot, N.; Tranchant, C.; Rudolf, G.; Chelly, J.; et al. Early-Onset Parkinsonism is a Manifestation of the PPP2R5D p.E200K Mutation. Ann. Neurol. 2020, 88, 1028–1033. [Google Scholar] [CrossRef]
- Stevens, S.J.C.; Ravenswaaij-Arts, C.M.A.; Janssen, J.W.H.; Klein Wassink-Ruiter, J.S.; van Essen, A.J.; Dijkhuizen, T.; van Rheenen, J.; Heuts-Vijgen, R.; Stegmann, A.P.A.; Smeets, E.E.J.G.L.; et al. MYT1L is a candidate gene for intellectual disability in patients with 2p25.3 (2pter) deletions. Am. J. Med. Genet. Part A 2011, 155A, 2739–2745. [Google Scholar] [CrossRef]
- De Ligt, J.; Willemsen, M.H.; van Bon, B.W.M.; Kleefstra, T.; Yntema, H.G.; Kroes, T.; Vulto-van Silfhout, A.T.; Koolen, D.A.; de Vries, P.; Gilissen, C.; et al. Diagnostic exome sequencing in persons with severe intellectual disability. N. Engl. J. Med. 2012, 367, 1921–1929. [Google Scholar] [CrossRef] [Green Version]
- De Rocker, N.; Vergult, S.; Koolen, D.; Jacobs, E.; Hoischen, A.; Zeesman, S.; Bang, B.; Bena, F.; Bockaert, N.; Bongers, E.M.; et al. Refinement of the critical 2p25.3 deletion region: The role of MYT1L in intellectual disability and obesity. Genet. Med. 2015, 17, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loid, P.; Makitie, R.; Costantini, A.; Viljakainen, H.; Pekkinen, M.; Makitie, O. A novel MYT1L mutation in a patient with severe early-onset obesity and intellectual disability. Am. J. Med. Genet. Part A 2018, 176A, 1972–1975. [Google Scholar] [CrossRef] [PubMed]
- Mayo, S.; Rosello, M.; Monfort, S.; Oltra, S.; Orellana, C.; Martinez, F. Haploinsufficiency of the MYT1L gene causes intellectual disability frequently associated with behavioral disorder. Genet. Med. 2015, 17, 683–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boland, M.J.; Nazor, K.L.; Tran, H.T.; Szücs, A.; Lynch, C.L.; Paredes, R.; Tassone, F.; Sanna, P.P.; Hagerman, R.J.; Loring, J.F. Molecular analyses of neurogenic defects in a human pluripotent stem cell model of fragile X syndrome. Brain 2017, 140, 582–598. [Google Scholar] [CrossRef]
- Kumari, D.; Sciascia, N.; Usdin, K. Small Molecules Targeting H3K9 Methylation Prevent Silencing of Reactivated FMR1 Alleles in Fragile X Syndrome Patient Derived Cells. Genes 2020, 11, 356. [Google Scholar] [CrossRef] [Green Version]
- Musumeci, S.A.; Colognola, R.M.; Ferri, R.; Gigli, G.L.; Petrella, M.A.; Sanfilippo, S.; Bergonzi, P.; Tassinari, C.A. Fragile-X syndrome: A particular epileptogenic EEG pattern. Epilepsia 1988, 29, 41–47. [Google Scholar] [CrossRef]
- Loesch, D.Z.; Huggins, R.M.; Hagerman, R.J. Phenotypic variation and FMRP levels in fragile X. Ment. Retard. Dev. Disabil. Res. Rev. 2004, 10, 31–41. [Google Scholar] [CrossRef]
- Faravelli, F.; Murdolo, M.; Marangi, G.; Bricarelli, F.D.; Di Rocco, M.; Zollino, M. Mother to son amplification of a small subtelomeric deletion: A new mechanism of familial recurrence in microdeletion syndromes. Am. J. Med. Genet. Part A 2007, 143A, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tabolacci, E.; Pomponi, M.G.; Remondini, L.; Pietrobono, R.; Orteschi, D.; Nobile, V.; Pucci, C.; Musto, E.; Pane, M.; Mercuri, E.M.; et al. Co-Occurrence of Fragile X Syndrome with a Second Genetic Condition: Three Independent Cases of Double Diagnosis. Genes 2021, 12, 1909. https://doi.org/10.3390/genes12121909
Tabolacci E, Pomponi MG, Remondini L, Pietrobono R, Orteschi D, Nobile V, Pucci C, Musto E, Pane M, Mercuri EM, et al. Co-Occurrence of Fragile X Syndrome with a Second Genetic Condition: Three Independent Cases of Double Diagnosis. Genes. 2021; 12(12):1909. https://doi.org/10.3390/genes12121909
Chicago/Turabian StyleTabolacci, Elisabetta, Maria Grazia Pomponi, Laura Remondini, Roberta Pietrobono, Daniela Orteschi, Veronica Nobile, Cecilia Pucci, Elisa Musto, Marika Pane, Eugenio M. Mercuri, and et al. 2021. "Co-Occurrence of Fragile X Syndrome with a Second Genetic Condition: Three Independent Cases of Double Diagnosis" Genes 12, no. 12: 1909. https://doi.org/10.3390/genes12121909
APA StyleTabolacci, E., Pomponi, M. G., Remondini, L., Pietrobono, R., Orteschi, D., Nobile, V., Pucci, C., Musto, E., Pane, M., Mercuri, E. M., Neri, G., Genuardi, M., Chiurazzi, P., & Zollino, M. (2021). Co-Occurrence of Fragile X Syndrome with a Second Genetic Condition: Three Independent Cases of Double Diagnosis. Genes, 12(12), 1909. https://doi.org/10.3390/genes12121909