
Novel In-Frame Deletion in HTRA1 Gene, Responsible for Stroke at a Young Age and Dementia—A Case Study

 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
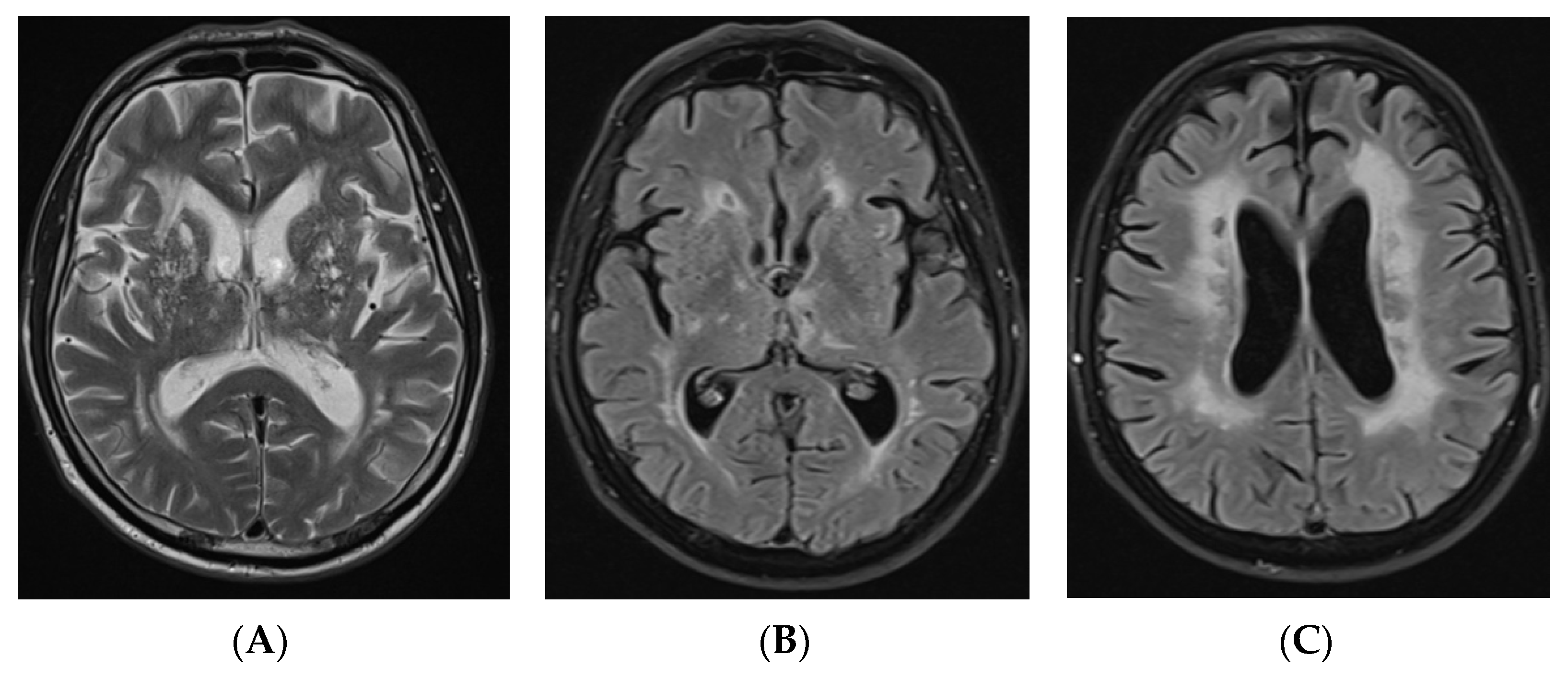
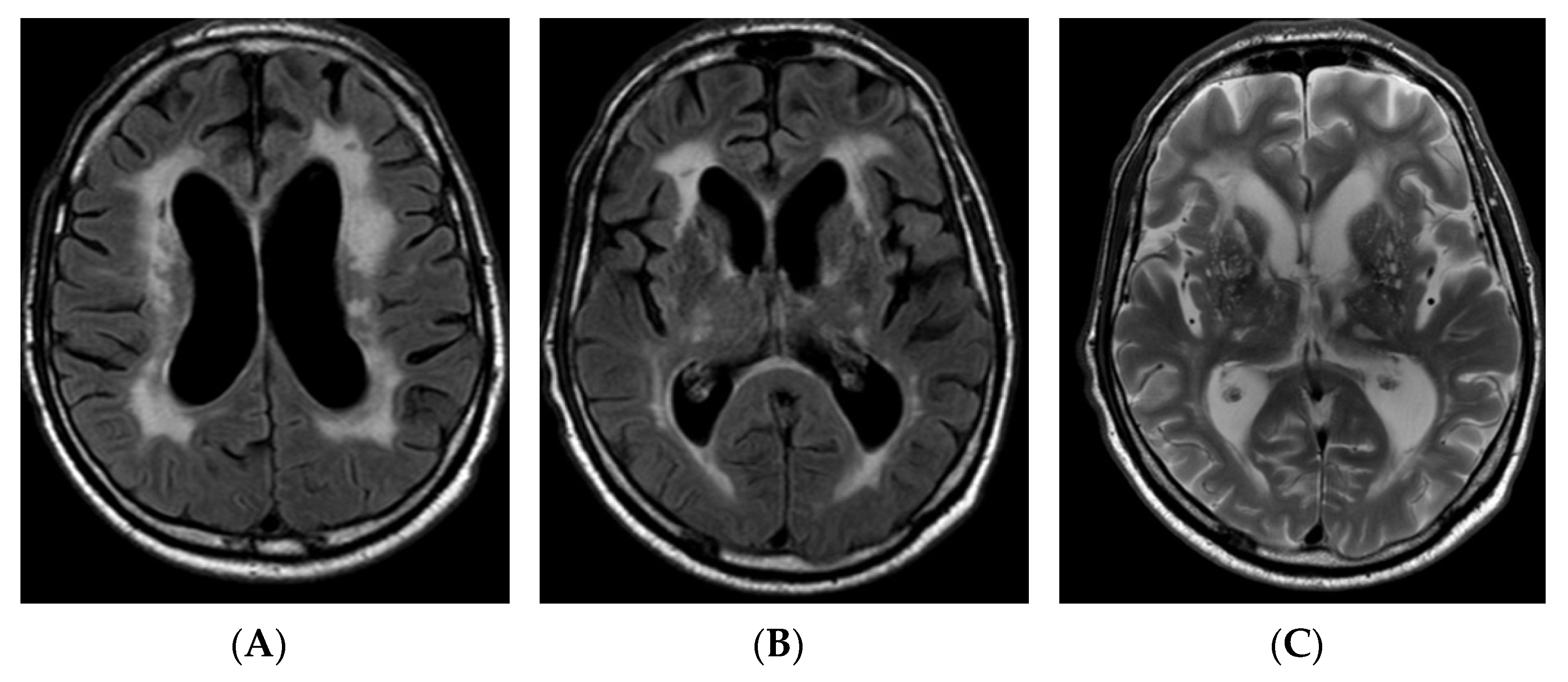
2.1. Case Report
2.2. Gene Sequencing
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pantoni, L. Cerebral small vessel disease: From pathogenesis and clinical characteristics to therapeutic challenges. Lancet Neurol. 2010, 9, 689–701. [Google Scholar] [CrossRef]
- Søndergaard, C.B.; Nielsen, J.E.; Hansen, C.K.; Christensen, H. Hereditary cerebral small vessel disease and stroke. Clin. Neurol. Neurosurg. 2017, 155, 45–57. [Google Scholar] [CrossRef]
- Verdura, E.; Hervé, D.; Scharrer, E.; del Mar Amador, M.; Guyant-Maréchal, L.; Philippi, A.; Corlobé, A.; Bergametti, F.; Gazal, S.; Prieto-Morin, C.; et al. Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain 2015, 138, 2347–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, K.; Shiga, A.; Fukutake, T.; Nozaki, H.; Miyashita, A.; Yokoseki, A.; Kawata, H.; Koyama, A.; Arima, K.; Takahashi, T.; et al. Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N. Engl. J. Med. 2009, 360, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-C.; Chung, C.-P.; Chao, N.-C.; Fuh, J.-L.; Chang, F.-C.; Soong, B.-W.; Liao, Y.-C. Characterization of Heterozygous HTRA1 Mutations in Taiwanese Patients with Cerebral Small Vessel Disease. Stroke 2018, 49, 1593–1601. [Google Scholar] [CrossRef] [PubMed]
- Onodera, O.; Nozaki, H.; Fukutake, T. HTRA1 Disorder [Internet]. GeneReviews® [Internet]. Available online: https://www.ncbi.nlm.nih.gov/books/NBK32533/ (accessed on 10 October 2021).
- Mendioroz, M.; Fernández-Cadenas, I.; del Río-Espinola, A.; Rovira, A.; Solé, E.; Fernández-Figueras, M.T.; García-Patos, V.; Sastre-Garriga, J.; Domingues-Montanari, S.; Álvarez-Sabín, J.; et al. A missense HTRA1 mutation expands CARASIL syndrome to the Caucasian population. Neurology 2010, 75, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, Z.; Meng, S.; Li, L.; Yang, H.; Zhang, X. A novel mutation of the high-temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). J. Int. Med. Res. 2013, 41, 1445–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OMIM. Phenotypic Series—PS125310. n.d. Available online: https://www.omim.org/phenotypicSeries/PS125310 (accessed on 15 August 2021).
- Nozaki, H.; Kato, T.; Nihonmatsu, M.; Saito, Y.; Mizuta, I.; Noda, T.; Koike, R.; Miyazaki, K.; Kaito, M.; Ito, S.; et al. Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology 2016, 86, 1964–1974. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, H.; Nishizawa, M.; Onodera, O. Features of cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Stroke 2014, 45, 3447–3453. [Google Scholar] [CrossRef]
- Uemura, M.; Nozaki, H.; Kato, T.; Koyama, A.; Sakai, N.; Ando, S.; Kanazawa, M.; Hishikawa, N.; Nishimoto, Y.; Polavarapu, K.; et al. HTRA1-related cerebral small vessel disease: A review of the literature. Front. Neurol. 2020, 11, 545. [Google Scholar] [CrossRef]
- GRCh38.p13—Genome-Assembly-NCBI n.d. Available online: https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.39/ (accessed on 18 October 2021).
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 2015, 43, D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Durbin, R.M.; Altshuler, D.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, A.S.; De La Vega, F.M.; Donnelly, P.; Egholm, M.; et al. A map of human genome variation from population-scale sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- PubMed. n.d. Available online: https://pubmed.ncbi.nlm.nih.gov/ (accessed on 18 October 2021).
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The ensembl variant effect predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- Vcfanno: Fast, Flexible Annotation of Genetic Variants. n.d. Available online: https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0973-5 (accessed on 26 October 2021).
- Sequence Variant Nomenclature. n.d. Available online: https://varnomen.hgvs.org/ (accessed on 27 October 2021).
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef] [PubMed]
- Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. HTRA proteases: Regulated proteolysis in protein quality control. Nat. Rev. Mol. Cell Biol. 2011, 12, 152–162. [Google Scholar] [CrossRef]
- Truebestein, L.; Tennstaedt, A.; Mönig, T.; Krojer, T.; Canellas, F.; Kaiser, M.; Clausen, T.; Ehrmann, M. Substrate-induced remodeling of the active site regulates human HTRA1 activity. Nat. Struct. Mol. Biol. 2011, 18, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Di Donato, I.; Bianchi, S.; Gallus, G.N.; Cerase, A.; Taglia, I.; Pescini, F.; Nannucci, S.; Battisti, C.; Inzitari, D.; Pantoni, L.; et al. Heterozygous mutations of HTRA1 gene in patients with familial cerebral small vessel disease. CNS Neurosci. Ther. 2017, 23, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuo, Z.; Cong, L.; Zhang, J.; Zhao, X. A novel heterozygous HTRA1 mutation is associated with autosomal dominant hereditary cerebral small vessel disease. Mol. Genet. Genomic Med. 2020, 8, e1111. [Google Scholar] [CrossRef] [PubMed]
- Ziaei, A.; Xu, X.; Dehghani, L.; Bonnard, C.; Zellner, A.; Jin Ng, A.Y.; Tohari, S.; Venkatesh, B.; Haffner, C.; Reversade, B.; et al. Novel mutation in HTRA1 in a family with diffuse white matter lesions and inflammatory features. Neurol. Genet. 2019, 5, e345. [Google Scholar] [CrossRef] [Green Version]
- VCV000995062.1—ClinVar-NCBI. n.d. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/995062/ (accessed on 18 October 2021).
- Liu, J.-Y.; Zhu, Y.-C.; Zhou, L.-X.; Wei, Y.-P.; Mao, C.-H.; Cui, L.-Y.; Peng, B.; Yao, M. HTRA1-related autosomal dominant cerebral small vessel disease. Chin. Med. J. (Engl.) 2020, 134, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Qin, X.; Shi, Y.; Gao, X.; Wang, F.; Wang, H.; Shang, J.; Zhao, J.; Zhang, J.; Shao, F. Genotype-phenotype correlations of heterozygous HTRA1-related cerebral small vessel disease: Case report and systematic review. Neurogenetics 2021, 22, 187–194. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grigaitė, J.; Šiaurytė, K.; Audronytė, E.; Preikšaitienė, E.; Burnytė, B.; Pranckevičienė, E.; Ekkert, A.; Utkus, A.; Jatužis, D. Novel In-Frame Deletion in HTRA1 Gene, Responsible for Stroke at a Young Age and Dementia—A Case Study. Genes 2021, 12, 1955. https://doi.org/10.3390/genes12121955
Grigaitė J, Šiaurytė K, Audronytė E, Preikšaitienė E, Burnytė B, Pranckevičienė E, Ekkert A, Utkus A, Jatužis D. Novel In-Frame Deletion in HTRA1 Gene, Responsible for Stroke at a Young Age and Dementia—A Case Study. Genes. 2021; 12(12):1955. https://doi.org/10.3390/genes12121955
Chicago/Turabian StyleGrigaitė, Julija, Kamilė Šiaurytė, Eglė Audronytė, Eglė Preikšaitienė, Birutė Burnytė, Erinija Pranckevičienė, Aleksandra Ekkert, Algirdas Utkus, and Dalius Jatužis. 2021. "Novel In-Frame Deletion in HTRA1 Gene, Responsible for Stroke at a Young Age and Dementia—A Case Study" Genes 12, no. 12: 1955. https://doi.org/10.3390/genes12121955
APA StyleGrigaitė, J., Šiaurytė, K., Audronytė, E., Preikšaitienė, E., Burnytė, B., Pranckevičienė, E., Ekkert, A., Utkus, A., & Jatužis, D. (2021). Novel In-Frame Deletion in HTRA1 Gene, Responsible for Stroke at a Young Age and Dementia—A Case Study. Genes, 12(12), 1955. https://doi.org/10.3390/genes12121955