
Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites


Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Sampling and DNA Sequencing
2.2. Assembly and Annotation of Mitogenomes
2.3. Identification of Repeats and Repeat-Mediated Homologous Recombinations
2.4. RNA Extraction and Sequencing
2.5. Identification of RNA Editing Sites in Mitochondrial Protein-Coding Genes (PCGs)
3. Results
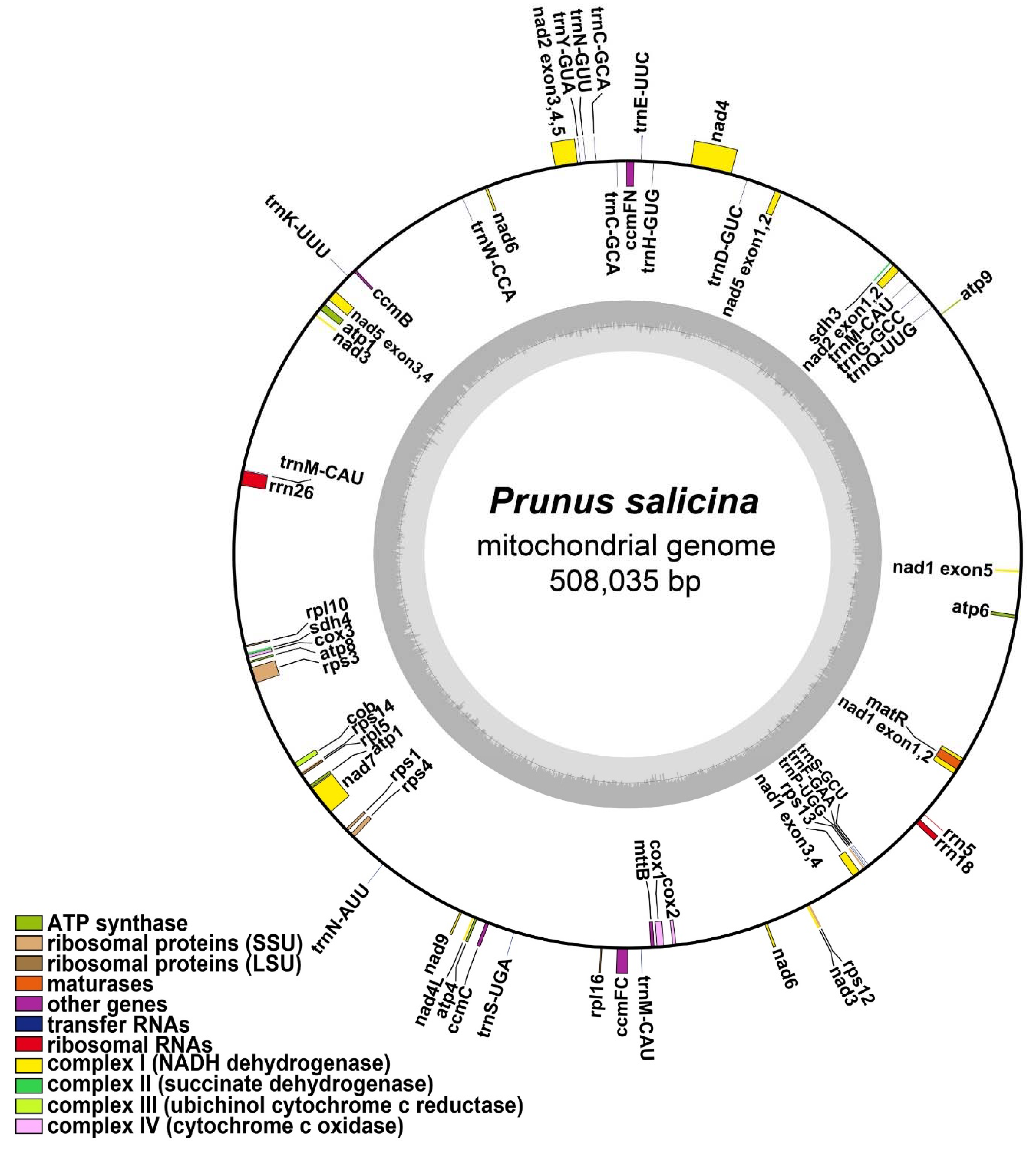
3.1. Characteristics of the P. salicina Mitogenome
3.2. Comparison of Mitochondrial Genomes in ROSACEAE
3.3. Repeats and Homologous Recombinations
3.4. Characteristics of RNA Editing Sites in Mitochondrial PCGs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lara, M.V.; Bonghi, C.; Famiani, F.; Vizzotto, G.; Walker, R.P.; Drincovich, M.F. Stone Fruit as Biofactories of Phytochemicals with Potential Roles in Human Nutrition and Health. Front. Plant Sci. 2020, 11, 562252. [Google Scholar] [CrossRef]
- Huang, Z.; Shen, F.; Chen, Y.; Cao, K.; Wang, L. Chromosome-scale genome assembly and population genomics provide insights into the adaptation, domestication, and flavonoid metabolism of Chinese plum. Plant. J. Cell Mol. Biol. 2021, 108, 1174–1192. [Google Scholar] [CrossRef]
- Noratto, G.D.; Garcia-Mazcorro, J.F.; Markel, M.; Martino, H.S.; Minamoto, Y.; Steiner, J.M.; Byrne, D.; Suchodolski, J.S.; Mertens-Talcott, S.U. Carbohydrate-Free Peach (Prunus persica) and Plum (Prunus salicina) Juice Affects Fecal Microbial Ecology in an Obese Animal Model. PLoS ONE 2014, 9, e101723. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Pengfei, W.; Brennan, H.; Ping, Q.; Bingxiang, L.; Feiyan, Z.; Hongbo, C.; Haijiang, C. Diversity of carotenoid composition, sequestering structures and gene transcription in mature fruits of four Prunus species. Plant. Physiol. Biochem. 2020, 151, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, W.; You, B.; Yang, S.; Xian, W.; Deng, Y.; Huang, W.; Yang, R. Phenolic profiles, bioaccessibility and antioxidant activity of plum (Prunus Salicina Lindl). Food Res. Int. 2021, 143, 110300. [Google Scholar] [CrossRef]
- Alister, C.; Araya, M.; Becerra, K.; Volosky, C.; Saavedra, J.; Kogan, M. Industrial prune processing and its effect on pesticide residue concentrations. Food Chem. 2018, 268, 264–270. [Google Scholar] [CrossRef]
- Fanning, K.J.; Topp, B.; Russell, D.; Stanley, R.; Netzel, M. Japanese plums (Prunus salicina Lindl.) and phytochemicals--breeding, horticultural practice, postharvest storage, processing and bioactivity. J. Sci. Food Agric. 2014, 94, 2137–2147. [Google Scholar] [CrossRef]
- Hernández-Herrero, J.A.; Frutos, M.J. Degradation kinetics of pigment, colour and stability of the antioxidant capacity in juice model systems from six anthocyanin sources. Int. J. Food Sci. Technol. 2011, 46, 2550–2557. [Google Scholar] [CrossRef]
- Kim, D.O.; Padilla-Zakour, O.I. Jam Processing Effect on Phenolics and Antioxidant Capacity in Anthocyanin-rich Fruits: Cherry, Plum, and Raspberry. J. Food Sci. 2004, 69, S395–S400. [Google Scholar] [CrossRef]
- Fiol, A.; García-Gómez, B.E.; Jurado-Ruiz, F.; Alexiou, K.; Howad, W.; Aranzana, M.J. Characterization of Japanese Plum (Prunus salicina) PsMYB10 Alleles Reveals Structural Variation and Polymorphisms Correlating with Fruit Skin Color. Front. Plant Sci. 2021, 12, 655267. [Google Scholar] [CrossRef]
- Numaguchi, K.; Akagi, T.; Kitamura, Y.; Ishikawa, R.; Ishii, T. Interspecific introgression and natural selection in the evolution of Japanese apricot (Prunus mume). Plant. J. Cell Mol. Biol. 2020, 104, 1551–1567. [Google Scholar] [CrossRef]
- Głowacka, A.; Sitarek, M.; Rozpara, E.; Podwyszyńska, M. Pomological Characteristics and Ploidy Levels of Japanese Plum (Prunus salicina Lindl.) Cultivars Preserved in Poland. Plants 2021, 10, 884. [Google Scholar] [CrossRef] [PubMed]
- Mubarak, A.; Swinny, E.E.; Ching, S.Y.; Jacob, S.R.; Lacey, K.; Hodgson, J.M.; Croft, K.D. Considine MJ: Polyphenol composition of plum selections in relation to total antioxidant capacity. J. Agric. Food Chem. 2012, 60, 10256–10262. [Google Scholar] [CrossRef]
- Roger, A.J.; Muñoz-Gómez, S.A.; Kamikawa, R. The Origin and Diversification of Mitochondria. Curr. Biol. 2017, 27, R1177–R1192. [Google Scholar] [CrossRef] [Green Version]
- Berkowitz, O.; De Clercq, I.; Van Breusegem, F.; Whelan, J. Interaction between hormonal and mitochondrial signalling during growth, development and in plant defence responses. Plant. Cell Environ. 2016, 39, 1127–1139. [Google Scholar] [CrossRef]
- Ng, S.; De Clercq, I.; Van Aken, O.; Law, S.R.; Ivanova, A.; Willems, P.; Giraud, E.; Van Breusegem, F.; Whelan, J. Anterograde and retrograde regulation of nuclear genes encoding mitochondrial proteins during growth, development, and stress. Mol. Plant 2014, 7, 1075–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, L.W.; Guo, W.; Mower, J.P.; Palmer, J.D. High and Variable Rates of Repeat-Mediated Mitochondrial Genome Rearrangement in a Genus of Plants. Mol. Biol. Evol. 2018, 35, 2773–2785. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant. Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Kozik, A.; Rowan, B.A.; Lavelle, D.; Berke, L.; Schranz, M.E.; Michelmore, R.W.; Christensen, A.C. The alternative reality of plant mitochondrial DNA: One ring does not rule them all. PLoS Gen. 2019, 15, e1008373. [Google Scholar] [CrossRef] [Green Version]
- Christensen, A.C. Plant mitochondrial genome evolution can be explained by DNA repair mechanisms. Genome Biol. Evol. 2013, 5, 1079–1086. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, D.B. One ring to rule them all? Genome sequencing provides new insights into the ‘master circle’ model of plant mitochondrial DNA structure. New Phytol. 2013, 200, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Shang, L.; Zhu, Q.-H.; Fan, L.; Guo, L. Twenty years of plant genome sequencing: Achievements and challenges. Trends Plant. Sci. 2021. [CrossRef] [PubMed]
- O’Conner, S.; Li, L. Mitochondrial Fostering: The Mitochondrial Genome May Play a Role in Plant Orphan Gene Evolution. Front. Plant Sci. 2020, 11, 600117. [Google Scholar] [CrossRef]
- Guo, J.; Wang, P.; Cheng, Q.; Sun, L.; Wang, H.; Wang, Y.; Kao, L.; Li, Y.; Qiu, T.; Yang, W.; et al. Proteomic analysis reveals strong mitochondrial involvement in cytoplasmic male sterility of pepper (Capsicum annuum L.). J. Proteom. 2017, 168, 15–27. [Google Scholar] [CrossRef]
- Touzet, P.; Meyer, E.H. Cytoplasmic male sterility and mitochondrial metabolism in plants. Mitochondrion 2014, 19, 166–171. [Google Scholar] [CrossRef]
- Tan, Q.; Li, S.; Zhang, Y.; Chen, M.; Wen, B.; Jiang, S.; Chen, X.; Fu, X.; Li, D.; Wu, H.; et al. Chromosome-level genome assemblies of five Prunus species and genome-wide association studies for key agronomic traits in peach. Hortic. Res. 2021, 8, 213. [Google Scholar] [CrossRef]
- Yan, M.; Zhang, X.; Zhao, X.; Yuan, Z. The complete mitochondrial genome sequence of sweet cherry (Prunus avium cv. ‘summit’). Mitochondrial DNA Part B 2019, 4, 1996–1997. [Google Scholar] [CrossRef] [Green Version]
- Arseneau, J.R.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 2017, 17, 686–693. [Google Scholar] [CrossRef]
- Emerman, A.B.; Bowman, S.K.; Barry, A.; Henig, N.; Patel, K.M.; Gardner, A.F.; Hendrickson, C.L. NEBNext Direct: A Novel, Rapid, Hybridization-Based Approach for the Capture and Library Conversion of Genomic Regions of Interest. Curr. Protoc. Mol. Biol. 2017, 119, 7–30. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap and miniasm: Fast mapping and de novo assembly for noisy long sequences. Bioinformatics 2016, 32, 2103–2110. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [Green Version]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Misra, S.; Harris, N. Using Apollo to Browse and Edit Genome Annotations. Curr. Protoc. Bioinform. 2005, 12, 9.5.1–9.5.28. [Google Scholar] [CrossRef]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.P.; Lowe, T.M. tRNAscan-SE: Searching for tRNA Genes in Genomic Sequences. Methods Mol. Biol. 2019, 1962, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, D.; Green, P. Consed: A graphical editor for next-generation sequencing. Bioinformatics 2013, 29, 2936–2937. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Duan, N.; Sun, H.; Wang, N.; Fei, Z.; Chen, X. The complete mitochondrial genome sequence of Malus hupehensis var. pinyiensis. Mitochondrial DNA Part A DNA Mapp. Seq. Anal. 2016, 27, 2905–2906. [Google Scholar] [CrossRef]
- Chung, H.Y.; Won, S.Y.; Kang, S.H.; Sohn, S.H.; Kim, J.S. The complete mitochondrial genome of Wonwhang (Pyrus pyrifolia). Mitochondrial DNA Part B Resour. 2017, 2, 902–903. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Zhao, C.; Chen, F.; Liu, Y.; Zhang, S.; Wu, H.; Zhang, L.; Liu, Y. The complete mitochondrial genome of the early flowering plant Nymphaea colorata is highly repetitive with low recombination. BMC Genom. 2018, 19, 614. [Google Scholar] [CrossRef]
- Li, J.; Xu, Y.; Shan, Y.; Pei, X.; Yong, S.; Liu, C.; Yu, J. Assembly of the complete mitochondrial genome of an endemic plant, Scutellaria tsinyunensis, revealed the existence of two conformations generated by a repeat-mediated recombination. Planta 2021, 254, 36. [Google Scholar] [CrossRef]
- Wu, B.; Chen, H.; Shao, J.; Zhang, H.; Wu, K.; Liu, C. Identification of Symmetrical RNA Editing Events in the Mitochondria of Salvia miltiorrhiza by Strand-specific RNA Sequencing. Sci. Rep. 2017, 7, 42250. [Google Scholar] [CrossRef]
- He, Z.S.; Zhu, A.; Yang, J.B.; Fan, W.; Li, D.Z. Organelle Genomes and Transcriptomes of Nymphaea Reveal the Interplay between Intron Splicing and RNA Editing. Int. J. Mol. Sci. 2021, 22, 9842. [Google Scholar] [CrossRef]
- Takemura, M.; Oda, K.; Yamato, K.; Ohta, E.; Nakamura, Y.; Nozato, N.; Akashi, K.; Ohyama, K. Gene clusters for ribosomal proteins in the mitochondrial genome of a liverwort, Marchantia polymorpha. Nucleic Acids Res. 1992, 20, 3199–3205. [Google Scholar] [CrossRef] [Green Version]
- Vasupalli, N.; Kumar, V.; Bhattacharya, R.; Bhat, S.R. Analysis of mitochondrial recombination in the male sterile Brassica juncea cybrid Og1 and identification of the molecular basis of fertility reversion. Plant. Mol. Biol. 2021, 106, 109–122. [Google Scholar] [CrossRef]
- Wang, S.; Li, D.; Yao, X.; Song, Q.; Wang, Z.; Zhang, Q.; Zhong, C.; Liu, Y.; Huang, H. Evolution and Diversification of Kiwifruit Mitogenomes through Extensive Whole-Genome Rearrangement and Mosaic Loss of Intergenic Sequences in a Highly Variable Region. Genome Biol. Evol. 2019, 11, 1192–1206. [Google Scholar] [CrossRef]
- Varré, J.S.; D’Agostino, N.; Touzet, P.; Gallina, S.; Tamburino, R.; Cantarella, C.; Ubrig, E.; Cardi, T.; Drouard, L.; Gualberto, J.M.; et al. Complete Sequence, Multichromosomal Architecture and Transcriptome Analysis of the Solanum tuberosum Mitochondrial Genome. Int. J. Mol. Sci. 2019, 20, 4788. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.F.; Jansen, R.K.; Zanis, M.J.; Emery, N.C. Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Am. J. Bot. 2015, 102, 1751–1752. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Li, X.Q. Organellar genome copy number variation and integrity during moderate maturation of roots and leaves of maize seedlings. Curr. Genet. 2015, 61, 591–600. [Google Scholar] [CrossRef]
- Gyorfy, M.F.; Miller, E.R.; Conover, J.L.; Grover, C.E.; Wendel, J.F.; Sloan, D.B.; Sharbrough, J. Nuclear-cytoplasmic balance: Whole genome duplications induce elevated organellar genome copy number. Plant. J. Cell Mol. Biol. 2021, 108, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhang, Y.; Havey, M.J.; Shou, W. Copy numbers of mitochondrial genes change during melon leaf development and are lower than the numbers of mitochondria. Hortic. Res. 2019, 6, 95. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Cai, X.; Hu, S.; Li, Y.; Fan, Y.; Tan, S.; Liu, Q.; Zhou, W. Comparative Analysis of the Mitochondrial Genomes of Nicotiana tabacum: Hints Toward the Key Factors Closely Related to the Cytoplasmic Male Sterility Mechanism. Front. Genet. 2020, 11, 257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.F.; Zhao, K.; Lv, J.H.; Huo, J.L.; Wang, Z.R.; Wan, H.J.; Zhu, H.S.; Zhang, Z.Q.; Shao, G.F.; Wang, J.; et al. Orf165 is associated with cytoplasmic male sterility in pepper. Genet. Mol. Biol. 2021, 44, e20210030. [Google Scholar] [CrossRef]
- Quiñones, V.; Zanlungo, S.; Holuigue, L.; Litvak, S.; Jordana, X. The cox1 initiation codon is created by RNA editing in potato mitochondria. Plant. Physiol. 1995, 108, 1327–1328. [Google Scholar] [CrossRef] [Green Version]
- Zanlungo, S.; Quiñones, V.; Holuigue, L.; Jordana, X. Identification of the RPS10 polypeptide encoded by the mitochondrial genome in Solanum tuberosum: Translation initiates at a genomic-encoded AUG and not at a conserved AUG codon created by RNA editing. Physiol. Plant. 2000, 110, 256–261. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Name of Genes | |
|---|---|---|
| Core genes | ATP synthase | atp1, atp4, atp6 1, atp8, atp9 |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFc 2, ccmFn | |
| Ubichinol cytochrome c reductase | cob | |
| Cytochrome c oxidase | cox1, cox2, cox3 | |
| Maturases | matR | |
| Transport membrane protein | mttB | |
| NADH dehydrogenase | nad1 2, nad2 2, nad3 (×2), nad4 2, nad4L, nad5 2, nad6 (×2), nad7 2, nad9 | |
| Variable genes | Large subunit of ribosome | rpl5, rpl10, rpl16 |
| Small subunit of ribosome | rps1, rps3 2, rps4, rps12, rps13, rps14 | |
| Succinate dehydrogenase | sdh3 1, sdh4 | |
| rRNA genes | Ribosomal RNAs | rrn5, rrn18, rrn26 |
| tRNA genes | Transfer RNAs | trnY-GUA, trnW-CCA 3, trnS-UGA, trnS-GCU, trnQ-UUG, trnP-UGG, trnN-GUU, trnN-AUU 3, trnM-CAU (×3), trnK-UUU, trnH-GUG, trnG-GCC, trnF-GAA, trnE-UUC, trnD-GUC, trnC-GCA (×2) |
| Species | Accession Number | Genome Size (bp) | GC Content (%) | Number of PCGs | Multicopy Genes | |
|---|---|---|---|---|---|---|
| Core Genes | Variable Genes | |||||
| Rosa chinensis | CM009589.1 | 313,448 | 45.48 | 24 | 9 (1) | rps3 |
| Fragaria orientalis | NC_057524.1 | 275,143 | 45.24 | 24 | 7 | |
| Malus domestica | NC_018554.1 | 396,947 | 45.39 | 24 | 9 (1) | rps12 |
| Malus hupehensis var. mengshanensis | KR534606.1 | 422,555 | 45.21 | 24 (2) | 9 (1) | atp8, cox2, rps3 |
| Eriobotrya japonica | NC_045228.1 | 434,980 | 45.42 | 24 (1) | 10 | nad4 |
| Pyrus betulifolia | NC_054332.1 | 469,928 | 45.28 | 24 (2) | 10 (2) | atp1, nad6, rps1, rps4 |
| Sorbus aucuparia | NC_052880.1 | 384,977 | 45.39 | 24 (1) | 9 | matR |
| Sorbus torminalis | NC_052879.1 | 386,758 | 45.31 | 24 | 9 | |
| Prunus avium | MT975322.1 | 444,582 | 45.62 | 24 | 10 (1) | rps3 |
| Prunus salicina | OK563724.1 | 508,035 | 45.43 | 24 (2) | 10 | nad3, nad6 |
| Repeat | Length (bp) | Direction | Position | Reads Support Master Circle Conformation | Reads Support Alternative Conformation |
|---|---|---|---|---|---|
| R1 | 511 | + | 200803–201313 | 182 | 11 |
| – | 419510–419000 | (94.30%) | (5.70%) | ||
| R2 | 472 | + | 156450–156921 | 167 | 4 |
| – | 410364–409893 | (96.67%) | (2.33%) | ||
| R3 | 393 | + | 164678–165070 | 135 | 4 |
| – | 345427–345035 | (97.12%) | (2.88%) | ||
| R4 | 385 | + | 361588–361972 | 166 | 3 |
| + | 501441–501825 | (98.22%) | (1.78%) | ||
| R5 | 299 | + | 280928–281226 | 163 | 4 |
| – | 358365–358067 | (97.60%) | (2.40%) | ||
| R6 | 294 | + | 123383–123676 | 149 | 5 |
| – | 154864–154571 | (96.75%) | (3.25%) | ||
| R7 | 195 | + | 215708–215902 | 176 | 5 |
| + | 303436–303630 | (97.24%) | (2.76%) | ||
| R8 | 175 | + | 290959–291133 | 182 | 1 |
| + | 362216–362390 | (99.45%) | (0.55%) | ||
| R9 | 111 | + | 89335–89445 | 171 | 1 |
| – | 343225–343115 | (99.42%) | (0.58%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fang, B.; Li, J.; Zhao, Q.; Liang, Y.; Yu, J. Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites. Genes 2021, 12, 1970. https://doi.org/10.3390/genes12121970
Fang B, Li J, Zhao Q, Liang Y, Yu J. Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites. Genes. 2021; 12(12):1970. https://doi.org/10.3390/genes12121970
Chicago/Turabian StyleFang, Bo, Jingling Li, Qian Zhao, Yuping Liang, and Jie Yu. 2021. "Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites" Genes 12, no. 12: 1970. https://doi.org/10.3390/genes12121970
APA StyleFang, B., Li, J., Zhao, Q., Liang, Y., & Yu, J. (2021). Assembly of the Complete Mitochondrial Genome of Chinese Plum (Prunus salicina): Characterization of Genome Recombination and RNA Editing Sites. Genes, 12(12), 1970. https://doi.org/10.3390/genes12121970