
Microarray Genotyping Identifies New Loci Associated with Dementia in Parkinson’s Disease
by
 ,
,
Sungyang Jo
1 ,
,
Kye Won Park
2,
Yun Su Hwang
1,
Seung Hyun Lee
1,
Ho-Sung Ryu
3 and
Sun Ju Chung
1,* 1
Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul 05505, Korea
2
Department of Neurology, Uijeongbu Eulji Medical Center, Eulji University School of Medicine, Uijeongbu-si 11759, Gyeonggi-do, Korea
3
Department of Neurology, Kyungpook National University Hospital, Daegu 41944, Korea
*
Author to whom correspondence should be addressed.
Genes 2021, 12(12), 1975; https://doi.org/10.3390/genes12121975
Submission received: 22 October 2021
/
Revised: 30 November 2021
/
Accepted: 2 December 2021
/
Published: 10 December 2021
(This article belongs to the Special Issue Parkinson's Disease: Genetics and Pathogenesis)
Abstract
:Dementia is one of the most disabling nonmotor symptoms of Parkinson’s disease (PD). However, the risk factors contributing to its development remain unclear. To investigate genetic variants associated with dementia in PD, we performed microarray genotyping based on a customized platform utilizing variants identified in previous genetic studies. Microarray genotyping was performed in 313 PD patients with dementia, 321 PD patients without dementia, and 635 healthy controls. The primary analysis was performed using a multiple logistic regression model adjusted for age and sex. SNCA single nucleotide polymorphism (SNP) rs11931074 was determined to be most significantly associated with PD (odds ratio = 0.66, 95% confidence interval = 0.56–0.78, p = 7.75 × 10−7). In the analysis performed for patients with PD only, MUL1 SNP rs3738128 (odds ratio = 2.52, 95% confidence interval = 1.68–3.79, p = 8.75 × 10−6) was found to be most significantly associated with dementia in PD. SNPs in ZHX2 and ERP29 were also associated with dementia in PD. This microarray genomic study identified new loci of MUL1 associated with dementia in PD, suggesting an essential role of mitochondrial dysfunction in the development of dementia in patients with PD.
1. Introduction
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease globally, affecting more than six million people worldwide [1]. The diagnosis of PD is based on specific motor symptoms, including bradykinesia, rigidity, tremor, or gait disturbance [2]. However, patients with PD suffer from various nonmotor symptoms, such as fatigue, pain, sleep disturbance, dementia, depression, anxiety, and autonomic dysfunction [3]. Dementia is one of the disabling nonmotor symptoms that substantially impairs the quality of life of patients with PD, increasing caregiver burden and economic costs [4]. The prevalence of dementia is high, with up to 75% of patients with PD developing dementia within 10 years from diagnosis [5,6]. However, the determining factors involved in the development of dementia in patients with PD are still unclear.
Genome-wide association studies (GWAS) have widened our understanding of the genetics of PD and have identified more than 90 genetic loci that are associated with the development of PD [7,8,9,10,11,12,13]. However, a majority of the previously conducted GWAS have focused on the susceptibility of PD, and GWAS specifically investigating motor or nonmotor presentations—including dementia—of PD have been limited. In a recent GWAS, we reported that RYR2 and other genetic loci are associated with cognitive impairment in PD; however, the assessment of cognitive function was based only on Mini-Mental Status Examination (MMSE) and the Montreal Cognitive Assessment (MoCA) scores [14]. Many recent GWAS have reported genomic variants, including GBA, APOE, SNCA, and CNTN1 that are associated with dementia with Lewy bodies (DLB) [15,16,17,18]. Although Parkinson’s disease dementia (PDD) and DLB share clinical, neurochemical, and morphological features, no consensus has been established yet with respect to the consideration of the two extremes on the one continuous spectrum of Lewy body disease [19]. Interestingly, in a large multinational cohort of patients with PD, PDD, and DLB, parkinsonism and dementia showed two distinct association profiles with the 3′ or 5′ regions of the SNCA gene, suggesting that PD, PDD, and DLB have distinct genetic etiologies. Therefore, further studies undertaking genome-wide investigations are necessary to identify distinct genetic variants associated with the development of dementia in patients with PD, independent of DLB.
In this study, we employed a novel customized microarray platform to comprehensively investigate the genetic variants associated with dementia in patients with PD.
2. Materials and Methods
2.1. Study Population
We prospectively enrolled patients with PDD, patients with PD without dementia (PD-ND), and healthy controls at Asan Medical Center, Seoul, Korea. All participants were ethnic Koreans. The diagnosis of PD was based on the UK Brain Bank criteria [2] and the diagnosis of PDD was based on the criteria proposed by the Movement Disorder Society Task Force [20]. Healthy controls were recruited from among the spouses of the patients, and the inclusion criterion included the absence of neurological diseases including PD or dementia. Blood samples were collected from all participants for genetic tests, and patient information including that related to age, sex, and educational qualification (number of years of education) was collected at the time of sampling. Mini-Mental Status Examination (MMSE) was performed for the screening of cognitive function. For patients with PD, age at disease onset, age at diagnosis of dementia if applicable, age at latest follow-up, and the latest MMSE scores were obtained.
2.2. Development of Microarray Genotyping Platform
We designed a microarray genotyping platform that contained genetic variants with biological plausibility for PDD, suggested by our previous GWAS or other previous genetic studies. The platform included: (1) Genetic variants that showed a high level of association (p-value < 10−4) with PD in our previous GWAS performed using ethnicity-specific Korean Chip (K-CHIP). K-CHIP was designed by the Center for Genome Science, Korea National Institute of Health (4845–301, 3000–3031) (www.cdc.go.kr) [14,21]. K-CHIP consists of an imputation GWAS grid (505,000 Asian-based grid with minor allele frequency (MAF) > 5% in Asians); exome content (84,000 Korean-based grid with MAF > 5% in Koreans, 149,000 coding single-nucleotide polymorphisms, and insertions and deletions determined based on data derived from 2000 whole-exome sequences and 400 whole-genome sequences with MAF > 0.1%); new exome/loss of function contents (44,000 variants); expression quantitative trait loci (17,000 variants); genes associated with absorption, distribution, metabolism, and excretion; and other miscellaneous variants. (2) Genetic variants that showed significant association with PD in previous GWAS [7,8,9,10,11,12,13]. (3) Genetic mutations that were reported to be a cause of monogenic familial PD with Mendelian inheritance (https://www.omim.org/). (4) Genetic variants that showed significant association with DLB in previous GWAS [15,16,22]. (5) Genetic variants that showed significant association with Alzheimer’s disease in previous GWAS [23,24,25,26]. (6) Genetic variants associated with neuroinflammation in previous GWAS [11,27,28].
Annotation of the variants was performed using the nspEff tool to confirm the distribution of the gene effect [29]. From a total of 219,065 variants, we excluded 109,804 “novel—not recommended and neutral” markers for the score data, because the performance or efficacy of genotyping might be low (Table S1). The final selection was performed by excluding duplicate markers, markers not included in the 1000 genome project phase 3 data, markers with a minor allele frequency of zero in East Asian GWAS data, and proxy single nucleotide polymorphisms (SNPs) (tagging r2 > 0.8) (Table S2). The final candidate markers consisted of 74,224 markers (Table S3).
2.3. Sample Quality Control
Samples with a low call rate and high heterozygosity were excluded. Samples that deviated from the whole sample were excluded from the analysis by an assessment performed using multidimensional scaling. We also excluded excessive singleton, samples with gender discrepancies, and cryptic first-degree relatives using the PLINK program (version 1.90, NIH–NIDDK Laboratory of Biological Modeling, Bethesda, MD, USA).
2.4. SNP Quality Control
We performed an SNPolisher analysis to exclude low-quality SNPs. SNPs with call rates over 95% in both cases and controls were included. SNPs with p-value > 10−4 in a Hardy–Weinberg equilibrium test were excluded. We excluded SNPs with minor allele frequency < 1% in both cases and controls. We performed cluster quality control for every SNP with p < 0.001 using linkage disequilibrium within 150 kilobases through visual inspection.
2.5. Statistical Analysis
We compared the demographics and clinical characteristics of patients with PDD, those with PD-ND, and healthy controls using Kruskal-Wallis tests for continuous variables, which did not meet the assumption of the homogeneity of variance, as well as with chi-squared test for categorical variables. Post hoc analysis was performed using Dunnett’s post hoc tests and Bonferroni correction.
The association between the genetic variants and PD or PDD was analyzed using a multiple logistic regression model after adjusting for age, sex, and education years. For each genetic variant, we calculated the odds ratios (OR), 95% confidence interval (CI), and two-tailed p-value. Bonferroni correction was applied to adjust for multiple comparisons. Manhattan plots and quantile–quantile plots (Q-Q plots) were constructed for p-values of all genotyped variants that passed quality control.
Statistical analysis was performed using R (version 3.1.2, Free Software Foundation, Inc., Boston, MA, USA), the PLINK program (version 1.90, NIH–NIDDK Laboratory of Biological Modeling, Bethesda, MD, USA), Haploview (version 4.2, Daly Lab at the Broad Institute, Cambridge, MA, USA), and LocusZoom (version 1.4, University of Michigan, Department of Biostatistics, Center for Statistical Genetics, Ann Arbor, MI, USA).
3. Results
3.1. Clinical Characteristics
We enrolled 318 patients with PDD, 326 patients with PD-ND, and 648 healthy controls. After quality control assessment, 5 patients with PDD, 5 patients with PD-ND, and 13 healthy controls were excluded. The final study population included 313 patients with PDD, 321 patients with PD-ND, and 635 healthy controls. The ages noted at the latest follow-up for patients with PD or those noted at study enrollment for healthy controls were significantly different among the three groups (median 76.0 vs. 75.0 vs. 68.0, p < 0.001) (Table 1). In the post hoc analysis, ages noted at the latest follow-up were significantly lower among healthy controls than those among patients with PDD or PD-ND (all p < 0.001). The ages at disease onset and the disease durations were not significantly different between patients with PDD and PD-ND. The median disease duration was 12.0 years for both PDD and PD-ND groups. The percentage of females was significantly higher in the PDD group compared to that in the healthy controls (57.2% vs. 44.9%, p = 0.0007 in post hoc analysis). The number of education years (total years of academic education) was significantly lower in the PDD group than that in the PD-ND or healthy control group (both p < 0.001 in post hoc analysis).
3.2. Genetic Association with Susceptibility to PD
The 41,534 genetic variants that passed quality control were genotyped and analyzed. Multiple logistic regression with additive coding schemes was performed to compare genetic variants between patients with PD (both, patients with PDD and those with PD-ND) and healthy controls after adjusting for age and sex. Q-Q plots were generated for the diagnosis of patients with PD in comparison with healthy controls (Figure S1). The Manhattan plot is depicted in Figure 1. Among the top 10 genetic variants associated with PD, five SNPs were observed in the loci of SNCA (rs11931074, rs12642514, rs75876872, rs80184884, and rs75231811) (Table 2), and two SNCA SNPs (rs11931074 and rs12642514) showed statistical significance after Bonferroni correction (Figure 1). Among the SNCA SNPs, SNP rs11931074 was most significantly associated with PD (OR = 0.66, 95% CI = 0.56–0.78, p = 7.75 × 10–7). SPHK1 SNP rs2247856 (OR = 0.65, 95% CI = 0.53–0.80, p = 4.35 × 10–5) and FYN SNP rs7772036 (OR = 0.72, 95% CI = 0.61–0.85, p = 9.74 × 10–5) were also associated with PD.
3.3. Genetic Association with Dementia in PD
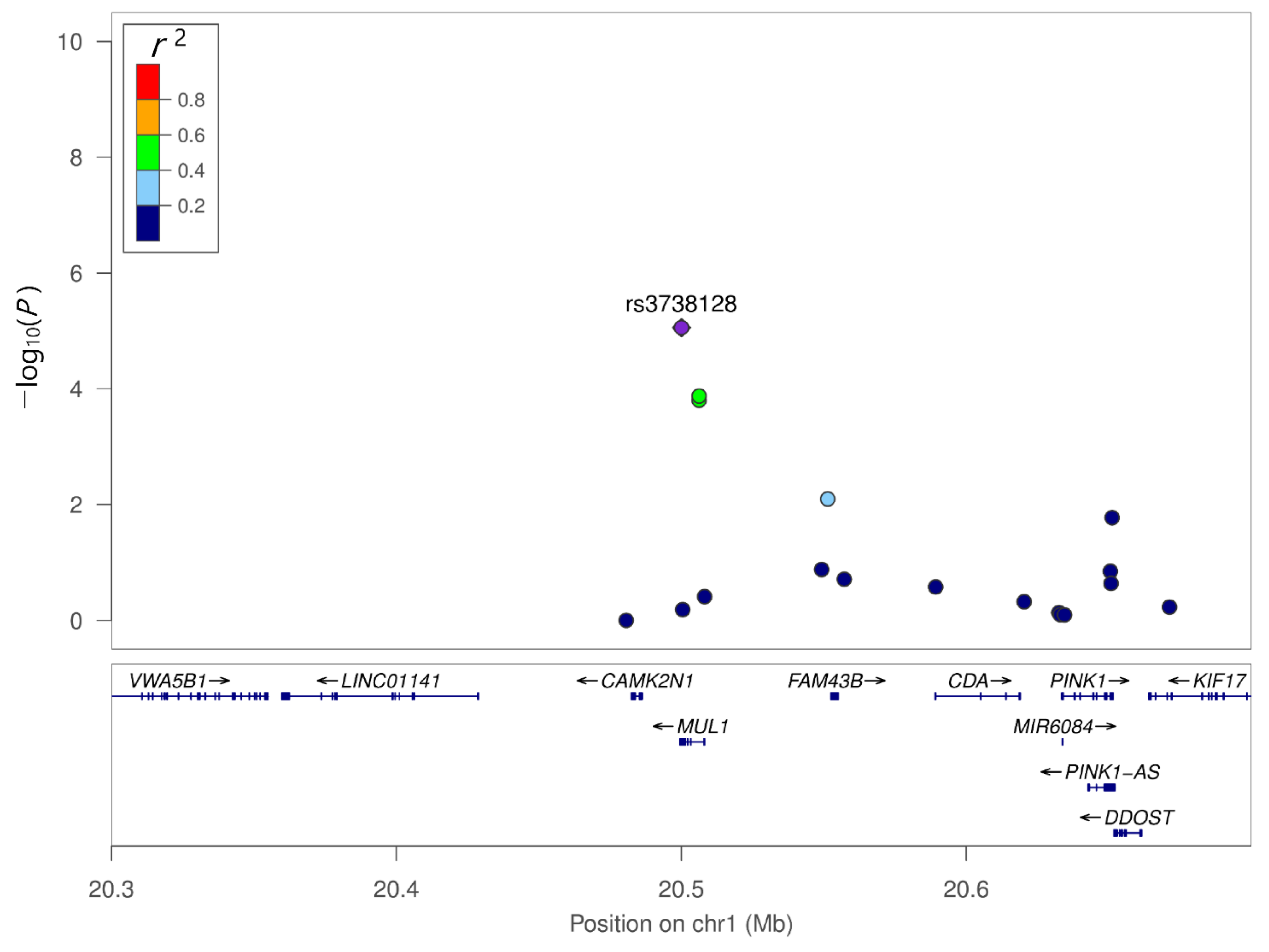
We compared genetic variants between PDD and PD-ND using multiple logistic regression with additive coding schemes after adjusting for age, sex, and education years. Q–Q plots were generated for the diagnosis of PDD compared with PD-ND (Figure S2). The respective Manhattan plot is depicted in Figure 2. Among the top 10 SNPs associated with PDD, two SNPs were observed in the loci of MUL1 (rs3738128 and rs12566937) (Table 3). MUL1 SNP rs3738128 (OR = 2.52, 95% CI = 1.68–3.79, p = 8.75 × 106) was most significantly associated with dementia in PD. In linkage analysis, MUL1 SNP rs12566937 showed moderate linkage disequilibrium with MUL1 SNP rs3738128, which was associated with the lowest p-value (Figure 3). SNPs in ZHX2 (OR = 0.56 95% CI = 0.43–0.74, p = 3.65 × 10–5) and ERP29 (OR = 3.05, 95% CI = 1.77–5.27, p = 6.41 × 10–5) were also associated with dementia in PD. However, following Bonferroni correction, none of the SNPs showed statistical significance.
4. Discussion
In this study, we identified genetic variants that were significantly associated with dementia in patients with PD with a median disease duration of over 12 years. The MUL1 SNP rs3738128 showed the most significant association with dementia in PD. ZHX2 and ERP29 also showed correlations with dementia in PD. The SNCA locus showed the most significant association with susceptibility to PD, consistent with the results of previous GWAS [7,8,9,10,11,12].
There were few studies investigating the role of MUL1 in the development of dementia in PD, and one case-control study conducted in China showed that MUL1 SNP rs529974 was correlated with the development of PD [30]. MUL1 encodes mitochondrial ubiquitin ligase 1, a mitochondrial E3 protein ligase that regulates mitofusin. The mitochondria are involved in cellular energy production and cell survival, playing an important role in the neurodegenerative process in PD [31]. Mitochondrial genes such as parkin, PINK1, DJ-1, LRRK2, ATP13A2, and VPS35 are associated with PD [32]. An experimental study showed that MUL1 suppressed the mitochondrial phenotype in PINK1/parkin mutant dopaminergic neuron, and the knockdown of MUL1 in parkin knockout mouse cortical neurons augmented mitochondrial damage [33]. Therefore, mutants with MUL1 and parkin mutations are employed in the development of animal models of PD [34]. MUL1 overexpression has been shown to reduce the degeneration of dopaminergic neurons and enhance motor activity in neurons of flies fed with rotenone [35]. MUL1 dysfunction renders dopaminergic neurons susceptible to mitochondrial damage. The loss of MUL1 function may be more prominent when other mitochondrial dysfunctions exist as well, as a result of genetic variants or environmental toxins. The lack of correlation of MUL1 with PD in this study may be explained by the adjunctive role of MUL1 in mitochondrial function.
Considering that the MUL1 pathway regulates mitochondrial damage in both dopaminergic and cortical neurons [33,36], defects in the MUL1 pathway might affect the cognitive decline in PD. However, little is known about the association between MUL1 and cognitive decline in PD or other neurodegenerative diseases that cause dementia. Mitochondrial dysfunction induces energy deficiency, intracellular calcium imbalance, and oxidative stress, leading to synaptic dysfunction and neuronal cell loss [37]. This mechanism explains how mitochondrial dysfunction mediates cognitive decline in neurodegenerative diseases, such as Alzheimer’s disease. Mitochondrial dysfunction is also prominent among patients with PD [38]. When MUL1 is downregulated, cortical neurons, as well as dopaminergic neurons, might become more susceptible to damage due to mitochondrial dysfunction, leading to the progression of cortical neuronal loss, synaptic dysfunction, and cognitive decline. In addition, recent studies have revealed that amyloid-beta and p-tau interact with mitochondrial proteins, resulting in increased mitochondrial fragmentation and reduced mitochondrial fusion in Alzheimer’s disease [39]. Similarly, pathogenic alpha-synuclein and amyloid-beta found in the brains of patients with PDD [40,41] might interact with MUL1, leading to mitochondrial dysfunction. The significant association of MUL1 with dementia in PD suggests the biological plausibility of the involvement of mitochondrial dysfunction in the development of dementia in PD. Further studies are needed to elucidate the exact pathogenic mechanisms underlying the involvement of MUL1 in the development of dementia in PD.
Other genetic variants associated with dementia in PD were located in the loci of ZHX2 and ERP29. Few clinical studies have investigated the role of ZHX2 and ERP29 in PD or dementia. The gene ZHX2 encodes zinc-finger and homeodomain protein 2 (ZHX2) that regulates transcription and neuronal differentiation [42]. Genetic variants of ZHX2 were found in two affected members of familial corticobasal degeneration, mutations of which were predicted to impair protein function [43]. Both corticobasal degeneration and PD are neurodegenerative diseases characterized by damage to cortical neurons and cognitive decline. Since ZHX2 is also associated with cortical neuronogenesis [42], it may be associated with the progression to dementia. ERP29 gene encodes a 29 kDa endoplasmic reticulum protein (ERp29), which is ubiquitously expressed in cells and regulates protein transport between the endoplasmic reticulum and Golgi apparatus [44]. ERp29 is involved in protein misfolding and mistrafficking [44,45], which are potent pathogenic features of PD and Alzheimer’s disease [46]. Given that endoplasmic reticulum stress is related to Lewy body dementia [47], it is possible that ERp29 mutation also induces cortical neuronal damage and is linked to the progression of dementia in patients with PD.
In our study, the SNCA SNP rs11931074 was most significantly associated with susceptibility to PD, which is consistent with previous results [7,9,10,11,12,48]. Mutations in the SNCA gene were first found in familial PD with autosomal dominant inheritance [49,50], and several SNPs across the SNCA locus were also linked to the increased risk for sporadic PD in multiple GWAS [7,8,9,10,11,12]. The SNCA gene encodes alpha-synuclein, which is the main component of Lewy bodies, the pathologic hallmark of PD. Interestingly, SNCA SNP rs11931074, which showed the most significant association with PD in this study also has a distinct relationship with PD based on race [48]. The presence of SNCA SNP rs11931074 increases the risk of PD, as demonstrated by the allele model, homozygote model, and recessive model developed for the Asian population, while the association was found to be true only in an allele model developed for the Caucasian population. These results support the quality of PD samples used in this study and might emphasize the role of SNCA SNP rs11931074 in the development of PD in the Asian population.
We found that SPHK1 and FYN SNPs were associated with PD. SPHK1 gene encodes sphingosine kinase 1 protein, which phosphorylates sphingosine into sphingosine-1-phosphate (S1P). S1P synthesized by SPHK1 exerts mitogenic and anti-apoptotic effects in an autocrine or paracrine manner [51]. The expression of sphingosine kinase 1 was downregulated in experimental models of PD, and inhibition of sphingosine kinase 1 decreases cell viability and enhances the production of reactive oxygen species [52]. FYN gene encodes the Fyc protein, which is a tyrosine phosphotransferase enzyme belonging to the Src family of nonreceptor tyrosine kinases. Fyc has been suggested to regulate alpha-synuclein phosphorylation, oxidative stress-induced dopaminergic neuronal death, and enhancement of neuroinflammation [53]. Therefore, both sphingosine kinase 1 protein and Fyc were suggested as potential therapeutic targets for PD [51,53], and our data support the protective effects of SPHK1 and FYN in PD.
The strength of this study is that we used clinical diagnosis of dementia based on the long-term follow-up of patients with PD. The prevalence of dementia in patients with PD is 17% at 5 years after diagnosis and 46−75% at 10 years after diagnosis [6,54]. Therefore, including PD patients with a short follow-up duration would misclassify them as having PD without dementia. A previous GWAS investigating the cognitive decline in PD included patients whose median follow-up duration was 4 years [55], and another GWAS assessed cognition using cross-sectional MMSE scores or MoCA scores [14].
This study has a few limitations. First, the sample size was relatively small, which may explain why genetic variants associated with dementia in patients with PD did not remain statistically significant after stringent Bonferroni correction. Second, the biological functions of the genetic variants were not validated. However, the experimental studies on SPHK1, FYN, MUL1, ZHX2, and ERP29 genes, as discussed above, might support the biological plausibility of the involvement of these genes in PD. Therefore, future functional studies are required to confirm our results.
5. Conclusions
This microarray genomic study identified the new loci of MUL1 associated with dementia in PD, suggesting an essential role of mitochondrial dysfunction in the development of this nonmotor symptom of PD.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes12121975/s1, Figure S1: Quantile–quantile plot for Parkinson’s disease, Figure S2: Quantile–quantile plot for dementia in Parkinson’s disease, Table S1: Characteristics of the markers used in the microarray, Table S2: Staged verification of the markers, Table S3: Additional selection according to selection priority.
Author Contributions
Conceptualization, S.J. and S.J.C.; methodology, S.J., K.W.P., H.-S.R., and S.J.C.; formal analysis, S.J., Y.S.H., and S.H.L.; investigation, S.J., K.W.P., Y.S.H., S.H.L., and H.-S.R.; writing—original draft preparation, S.J. and S.J.C.; writing—review and editing, S.J., H.-S.R., and S.J.C.; funding acquisition, S.J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by the Korea Healthcare Technology R&D Project, Ministry of Health and Welfare, Republic of Korea (grant number: HI19C0256).
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board of Asan Medical Center (2019–0533, 2019–04-22).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author. The data are not publicly available due to the privacy of the study population.
Acknowledgments
We appreciate the unlimited academic support from Yoon Kim, Chairman of Samyang Holdings Corporation, Seoul, Korea.
Conflicts of Interest
The authors declare no conflict of interest.
References
- GBD 2015 Neurological Disorders Collaborator Group. Global, regional, and national burden of neurological disorders during 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017, 16, 877–897. [Google Scholar] [CrossRef] [Green Version]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schapira, A.H.V.; Chaudhuri, K.R.; Jenner, P. Non-motor features of Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.F.; Larsen, J.P.; Tysnes, O.B.; Alves, G. Prognosis of mild cognitive impairment in early Parkinson disease: The Norwegian ParkWest study. JAMA Neurol. 2013, 70, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Kurz, M.W. The epidemiology of dementia associated with Parkinson disease. J. Neurol. Sci. 2010, 289, 18–22. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Mason, S.L.; Evans, J.R.; Foltynie, T.; Brayne, C.; Robbins, T.W.; Barker, R.A. The CamPaIGN study of Parkinson’s disease: 10-year outlook in an incident population-based cohort. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y.; et al. Identification of risk loci for Parkinson disease in Asians and comparison of risk between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simón-Sánchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D.; Paisan-Ruiz, C.; Lichtner, P.; Scholz, S.W.; Hernandez, D.G.; et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Hamza, T.H.; Zabetian, C.P.; Tenesa, A.; Laederach, A.; Montimurro, J.; Yearout, D.; Kay, D.M.; Doheny, K.F.; Paschall, J.; Pugh, E.; et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat. Genet. 2010, 42, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Nalls, M.A.; Pankratz, N.; Lill, C.M.; Do, C.B.; Hernandez, D.G.; Saad, M.; DeStefano, A.L.; Kara, E.; Bras, J.; Sharma, M.; et al. Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat. Genet. 2014, 46, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Chang, D.; Nalls, M.A.; Hallgrímsdóttir, I.B.; Hunkapiller, J.; van der Brug, M.; Cai, F.; Kerchner, G.A.; Ayalon, G.; International Parkinson’s Disease Genomics Consortium; 23andMe Research Team; et al. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat. Genet. 2017, 49, 1511–1516. [Google Scholar] [CrossRef] [PubMed]
- Park, K.W.; Jo, S.; Kim, M.S.; Jeon, S.R.; Ryu, H.S.; Kim, J.; Park, Y.M.; Koh, S.B.; Lee, J.H.; Chung, S.J. Genomic association study for cognitive impairment in Parkinson’s disease. Front. Neurol. 2020, 11, 579268. [Google Scholar] [CrossRef]
- Guerreiro, R.; Ross, O.A.; Kun-Rodrigues, C.; Hernandez, D.G.; Orme, T.; Eicher, J.D.; Shepherd, C.E.; Parkkinen, L.; Darwent, L.; Heckman, M.G.; et al. Investigating the genetic architecture of dementia with Lewy bodies: A two-stage genome-wide association study. Lancet Neurol. 2018, 17, 64–74. [Google Scholar] [CrossRef] [Green Version]
- Rongve, A.; Witoelar, A.; Ruiz, A.; Athanasiu, L.; Abdelnour, C.; Clarimon, J.; Heilmann-Heimbach, S.; Hernández, I.; Moreno-Grau, S.; de Rojas, I.; et al. GBA and APOE ε4 associate with sporadic dementia with Lewy bodies in European genome wide association study. Sci. Rep. 2019, 9, 7013. [Google Scholar] [CrossRef]
- Chia, R.; Sabir, M.S.; Bandres-Ciga, S.; Saez-Atienzar, S.; Reynolds, R.H.; Gustavsson, E.; Walton, R.L.; Ahmed, S.; Viollet, C.; Ding, J.; et al. Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 2021, 53, 294–303. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Dementia with Lewy bodies and Parkinson’s disease-dementia: Current concepts and controversies. J. Neural Transm. 2018, 125, 615–650. [Google Scholar] [CrossRef]
- Dubois, B.; Burn, D.; Goetz, C.; Aarsland, D.; Brown, R.G.; Broe, G.A.; Dickson, D.; Duyckaerts, C.; Cummings, J.; Gauthier, S.; et al. Diagnostic procedures for Parkinson’s disease dementia: Recommendations from the movement disorder society task force. Mov. Disord. 2007, 22, 2314–2324. [Google Scholar] [CrossRef]
- Ryu, H.S.; Park, K.W.; Choi, N.; Kim, J.; Park, Y.M.; Jo, S.; Kim, M.J.; Kim, Y.J.; Kim, J.; Kim, K.; et al. Genomic analysis identifies new loci associated with motor complications in Parkinson’s disease. Front. Neurol. 2020, 11, 570. [Google Scholar] [CrossRef] [PubMed]
- Orme, T.; Guerreiro, R.; Bras, J. The genetics of dementia with Lewy bodies: Current understanding and future directions. Curr. Neurol. Neurosci. Rep. 2018, 18, 67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Villegas-Llerena, C.; Phillips, A.; Garcia-Reitboeck, P.; Hardy, J.; Pocock, J.M. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr. Opin. Neurobiol. 2016, 36, 74–81. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Petersen, J.; Rossjohn, J.; Fugger, L. HLA variation and disease. Nat. Rev. Immunol. 2018, 18, 325–339. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [Green Version]
- Taximaimaiti, R.; Li, H. MUL1 gene polymorphisms and Parkinson’s disease risk. Acta Neurol. Scand. 2019, 139, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Malpartida, A.B.; Williamson, M.; Narendra, D.P.; Wade-Martins, R.; Ryan, B.J. Mitochondrial dysfunction and mitophagy in Parkinson’s disease: From mechanism to therapy. Trends Biochem. Sci. 2021, 46, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Grünewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Puri, R.; Yang, H.; Lizzio, M.A.; Wu, C.; Sheng, Z.H.; Guo, M. MUL1 acts in parallel to the PINK1/parkin pathway in regulating mitofusin and compensates for loss of PINK1/parkin. eLife 2014, 3, e01958. [Google Scholar] [CrossRef] [PubMed]
- Doktór, B.; Damulewicz, M.; Pyza, E. Effects of MUL1 and PARKIN on the circadian clock, brain and behaviour in Drosophila Parkinson’s disease models. BMC Neurosci. 2019, 20, 24. [Google Scholar] [CrossRef]
- Doktór, B.; Damulewicz, M.; Pyza, E. Overexpression of mitochondrial ligases reverses rotenone-induced effects in a Drosophila model of Parkinson’s disease. Front. Neurosci. 2019, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Puri, R.; Cheng, X.T.; Lin, M.Y.; Huang, N.; Sheng, Z.H. Mul1 restrains Parkin-mediated mitophagy in mature neurons by maintaining ER-mitochondrial contacts. Nat. Commun. 2019, 10, 3645. [Google Scholar] [CrossRef] [Green Version]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Reeve, A.K.; Grady, J.P.; Cosgrave, E.M.; Bennison, E.; Chen, C.; Hepplewhite, P.D.; Morris, C.M. Mitochondrial dysfunction within the synapses of substantia nigra neurons in Parkinson’s disease. NPJ Parkinson’s Dis. 2018, 4, 9. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuboi, Y.; Uchikado, H.; Dickson, D.W. Neuropathology of Parkinson’s disease dementia and dementia with Lewy bodies with reference to striatal pathology. Parkinsonism Relat. Disord. 2007, 13, S221–S224. [Google Scholar] [CrossRef]
- Smith, C.; Malek, N.; Grosset, K.; Cullen, B.; Gentleman, S.; Grosset, D.G. Neuropathology of dementia in patients with Parkinson’s disease: A systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1234–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Qiu, R.; Wang, J.; Zhang, H.; Murai, K.; Lu, Q. ZHX2 Interacts with Ephrin-B and regulates neural progenitor maintenance in the developing cerebral cortex. J. Neurosci. 2009, 29, 7404–7412. [Google Scholar] [CrossRef]
- Fekete, R.; Bainbridge, M.; Baizabal-Carvallo, J.F.; Rivera, A.; Miller, B.; Du, P.; Kholodovych, V.; Powell, S.; Ondo, W. Exome sequencing in familial corticobasal degeneration. Parkinsonism Relat. Disord. 2013, 19, 1049–1052. [Google Scholar] [CrossRef] [Green Version]
- Brecker, M.; Khakhina, S.; Schubert, T.J.; Thompson, Z.; Rubenstein, R.C. The probable, possible, and novel functions of ERp29. Front. Physiol. 2020, 11, 574339. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int. J. Mol. Sci. 2012, 14, 434–456. [Google Scholar] [CrossRef]
- Forloni, G.; Terreni, L.; Bertani, I.; Fogliarino, S.; Invernizzi, R.; Assini, A.; Ribizzi, G.; Negro, A.; Calabrese, E.; Volonté, M.A.; et al. Protein misfolding in Alzheimer’s and Parkinson’s disease: Genetics and molecular mechanisms. Neurobiol. Aging 2002, 23, 957–976. [Google Scholar] [CrossRef]
- Baek, J.H.; Whitfield, D.; Howlett, D.; Francis, P.; Bereczki, E.; Ballard, C.; Hortobágyi, T.; Attems, J.; Aarsland, D. Unfolded protein response is activated in Lewy body dementias. Neuropathol. Appl. Neurobiol. 2016, 42, 352–365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du, B.; Xue, Q.; Liang, C.; Fan, C.; Liang, M.; Zhang, Y.; Bi, X.; Hou, L. Association between alpha-synuclein (SNCA) rs11931074 variability and susceptibility to Parkinson’s disease: An updated meta-analysis of 41,811 patients. Neurol. Sci. 2020, 41, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. α-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef] [Green Version]
- Motyl, J.; Strosznajder, J.B. Sphingosine kinase 1/sphingosine-1-phosphate receptors dependent signalling in neurodegenerative diseases. The promising target for neuroprotection in Parkinson’s disease. Pharmacol. Rep. 2018, 70, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Pyszko, J.; Strosznajder, J.B. Sphingosine kinase 1 and sphingosine-1-phosphate in oxidative stress evoked by 1-methyl-4-phenylpyridinium (MPP+) in human dopaminergic neuronal cells. Mol. Neurobiol. 2014, 50, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Angelopoulou, E.; Paudel, Y.N.; Julian, T.; Shaikh, M.F.; Piperi, C. Pivotal role of Fyn Kinase in Parkinson’s disease and levodopa-induced dyskinesia: A novel therapeutic target? Mol. Neurobiol. 2021, 58, 1372–1391. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Batzu, L.; Halliday, G.M.; Geurtsen, G.J.; Ballard, C.; Chaudhuri, K.R.; Weintraub, D. Parkinson disease-associated cognitive impairment. Nat. Rev. Dis. Prim. 2021, 7, 47. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.M.X.; Lawton, M.A.; Jabbari, E.; Reynolds, R.H.; Iwaki, H.; Blauwendraat, C.; Kanavou, S.; Pollard, M.I.; Hubbard, L.; Malek, N.; et al. Genome-wide association studies of cognitive and motor progression in Parkinson’s disease. Mov. Disord. 2021, 36, 424–433. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Manhattan plots for Parkinson’s disease (PD). The genes nearest to the top 10 significant variants are labeled. The x-axis represents the base pair position of the variants from chromosome 1 to chromosome 22. The SNCA loci showed a statistically significant association with PD after Bonferroni correction. SNCA SNP rs11931074 was most significantly associated with PD (OR = 0.66, 95% CI = 0.56–0.78, p = 7.75 × 10–7). The SPHK1 and FYN loci were also associated with PD.
Figure 1.
Manhattan plots for Parkinson’s disease (PD). The genes nearest to the top 10 significant variants are labeled. The x-axis represents the base pair position of the variants from chromosome 1 to chromosome 22. The SNCA loci showed a statistically significant association with PD after Bonferroni correction. SNCA SNP rs11931074 was most significantly associated with PD (OR = 0.66, 95% CI = 0.56–0.78, p = 7.75 × 10–7). The SPHK1 and FYN loci were also associated with PD.

Figure 2.
Manhattan plots for dementia in Parkinson’s disease (PD). The genes nearest to the top 10 significant variants are labeled. The x-axis represents the base pair position of the variants from chromosome 1 to chromosome 22. The MUL1 loci was most significantly associated with dementia in PD. MUL1 SNP rs3738128 (OR = 2.52, 95% CI = 1.68–3.79, p = 8.75 × 10–6) was most significantly associated with dementia in PD. The ZHX2 and ERP29 loci were also associated with dementia in PD.
Figure 2.
Manhattan plots for dementia in Parkinson’s disease (PD). The genes nearest to the top 10 significant variants are labeled. The x-axis represents the base pair position of the variants from chromosome 1 to chromosome 22. The MUL1 loci was most significantly associated with dementia in PD. MUL1 SNP rs3738128 (OR = 2.52, 95% CI = 1.68–3.79, p = 8.75 × 10–6) was most significantly associated with dementia in PD. The ZHX2 and ERP29 loci were also associated with dementia in PD.

Figure 3.
Regional association plot of the genetic variants of MUL1. MUL1 SNP rs12566937 showed moderate linkage disequilibrium with MUL1 SNP rs3738128.
Figure 3.
Regional association plot of the genetic variants of MUL1. MUL1 SNP rs12566937 showed moderate linkage disequilibrium with MUL1 SNP rs3738128.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Baseline clinical characteristics of the study subjects.
| Characteristics | PD Dementia (N = 313) | PD without Dementia (N = 321) | Controls (N = 635) | p-Value |
|---|---|---|---|---|
| Age at onset, years | 64.0 (57.0−68.0) | 63.0 (57.0−68.0) | - | 0.449 |
| Age at latest follow-up, years | 76.0 (72.0–81.0) | 75.0 (72.0−80.0) | 68.0 (64.0−72.0) a,b | <0.001 |
| Disease duration, years | 12.0 (9.0−17.0) | 12.0 (9.0−16.0) | - | 0.896 |
| Female, N (%) | 179 (57.2%) | 166 (51.7%) | 285 (44.9%) a | 0.001 |
| Education, years | 6.0 (2.0−12.0) | 12.0 (6.0−16.0) c | 12.0 (9.0−16.0) a | <0.001 |
| Latest MMSE | 17.0(13.0−20.0) | 27.0 (26.0−29.0) c | 28.0 (26.0−29.0) a | <0.001 |
| Age at dementia, years | 73.0 (69.0−78.0) | - | - |
PD, Parkinson’s disease; MMSE, Mini-Mental Status Examination. a Significant difference compared with PD dementia using Dunn’s post hoc test. b Significant difference compared with PD without dementia using Dunn’s post hoc test. c Significant difference compared with healthy controls using Dunn’s post hoc test.
Table 2.
Top 10 genetic variants associated with Parkinson’s disease in the order of statistical significance.
Table 2.
Top 10 genetic variants associated with Parkinson’s disease in the order of statistical significance.
| Gene | SNP | Chr | Position | Region Relative to the Gene | Allele (Minor/Major) | Minor Allele Frequency (Case/Control) | OR (95% CI) | p-Value |
|---|---|---|---|---|---|---|---|---|
| SNCA, GPRIN3 | rs11931074 | 4 | 89718364 | intron, downstream, upstream | G/C | 0.37/0.46 | 0.66 (0.56, 0.78) | 7.75 × 10–7 |
| SNCA, GPRIN3 | rs12642514 | 4 | 89708246 | intron, downstream, upstream | A/C | 0.36/0.46 | 0.66 (0.58, 0.79) | 2.08 × 10–6 |
| SNCA | rs356191 | 4 | 89766969 | Intron | A/G | 0.06/0.10 | 0.52 (0.38, 0.70) | 2.64 × 10–5 |
| SNCA, GPRIN3 | rs80184884 | 4 | 89705068 | intron, downstream, upstream | G/A | 0.06/0.10 | 0.52 (0.38 0.71) | 4.24 × 10–5 |
| SPHK1 | rs2247856 | 17 | 76385474 | missense, UTR-5, exon | A/G | 0.16/0.22 | 0.65 (0.53, 0.80) | 4.35 × 10–5 |
| MYRIP | rs6599077 | 3 | 40055127 | Intron | A/G | 0.43/0.35 | 1.42(1.20, 1.68) | 4.81 × 10–5 |
| MRI100HG | rs577924 | 11 | 122264399 | Intron | C/T | 0.43/0.35 | 1.41 (1.19, 1.67) | 6.05 × 10–5 |
| SNCA, GPRIN3 | rs75876872 | 4 | 89705795 | intron, downstream, upstream | G/A | 0.05/0.08 | 0.49 (0.35, 0.69) | 6.07 × 10–5 |
| LOC339593 | rs1473702 | 20 | 11253884 | intron, downstream | C/T | 0.51/0.44 | 1.38 (1.18, 1.62) | 8.05 × 10–5 |
| FYN | rs7772036 | 6 | 111739596 | Intron | G/A | 0.32/0.39 | 0.72 (0.61, 0.85) | 9.74 × 10–5 |
Chr, chromosome; OR, odds ratio; CI, confidence interval.
Table 3.
Top 10 genetic variants associated with dementia in Parkinson’s disease in the order of statistical significance.
Table 3.
Top 10 genetic variants associated with dementia in Parkinson’s disease in the order of statistical significance.
| Gene | SNP | Chr | Position | Region Relative to the Gene | Allele (Minor/Major) | Minor Allele Frequency (Case/Control) | OR (95% CI) | p-Value |
|---|---|---|---|---|---|---|---|---|
| MUL1 | rs3738128 | 1 | 20499992 | UTR-3 | G/C | 0.07/0.11 | 2.52 (1.68, 3.79) | 8.75 × 10–6 |
| ZHX2 | rs11779459 | 8 | 122968311 | Intron | T/C | 0.34/0.29 | 0.56 (0.43, 0.74) | 3.65 × 10–5 |
| ERP29, NAA25 | rs4767293 | 12 | 112025492 | downstream | A/G | 0.04/0.06 | 3.05 (1.77, 5.27) | 6.41 × 10–5 |
| LINC01488 | rs7395791 | 11 | 69448148 | upstream, downstream | A/G | 0.56/0.50 | 0.61 (0.47, 0.78) | 8.44 × 10–5 |
| LINC01140 | rs7553864 | 1 | 87147675 | Intron | T/C | 0.14/0.19 | 1.88 (1.37, 2.6) | 1.15 × 10–4 |
| MUL1 | rs12566937 | 1 | 20506181 | Intron | G/T | 0.13/0.17 | 1.91 (1.37, 2.67) | 1.33 × 10–4 |
| LYZL1, C10orf126 | rs1889714 | 10 | 29099710 | upstream, downstream | A/G | 0.12/0.09 | 0.43 (0.28, 0.66) | 1.47 × 10–4 |
| AMY1C, LOC101928476, LOC100129138 | rs12026039 | 1 | 104028469 | downstream, upstream | G/A | 0.51/0.47 | 0.61 (0.47, 0.79) | 1.74 × 10–4 |
| DMRT1, KANK1 | rs912062 | 9 | 841152 | upstream, downstream | C/A | 0.17/0.22 | 1.76 (1.31, 2.37) | 1.82 × 10–4 |
| GLI2, LINC01101 | rs11688682 | 2 | 120590036 | Upstream | C/G | 0.08/0.04 | 2.62 (1.57, 4.37) | 2.30 × 10–4 |
SNP, single-nucleotide polymorphism; Chr, chromosome; OR, odds ratio; CI, confidence interval.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jo, S.; Park, K.W.; Hwang, Y.S.; Lee, S.H.; Ryu, H.-S.; Chung, S.J. Microarray Genotyping Identifies New Loci Associated with Dementia in Parkinson’s Disease. Genes 2021, 12, 1975. https://doi.org/10.3390/genes12121975
AMA Style
Jo S, Park KW, Hwang YS, Lee SH, Ryu H-S, Chung SJ. Microarray Genotyping Identifies New Loci Associated with Dementia in Parkinson’s Disease. Genes. 2021; 12(12):1975. https://doi.org/10.3390/genes12121975
Chicago/Turabian StyleJo, Sungyang, Kye Won Park, Yun Su Hwang, Seung Hyun Lee, Ho-Sung Ryu, and Sun Ju Chung. 2021. "Microarray Genotyping Identifies New Loci Associated with Dementia in Parkinson’s Disease" Genes 12, no. 12: 1975. https://doi.org/10.3390/genes12121975
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.