
The Mitochondrial Genome of a Freshwater Pelagic Amphipod Macrohectopus branickii Is among the Longest in Metazoa

 ,
,  , and
, and
Abstract
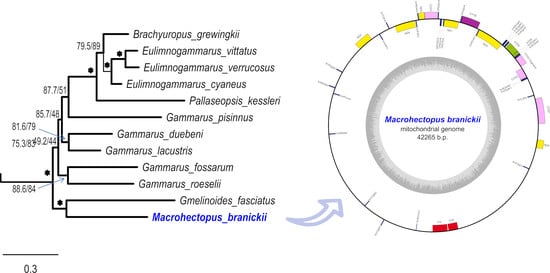
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Sequencing and Assembly
2.2. Mt Genome Sequence Verification and Annotation
2.3. Structural Analyses of M. branickii Mt Genome and Phylogenetic Inference of Amphipods
2.4. Statistical Analysis of Mt Genome Sequences from RefSeq
2.5. Phylogenetic Analysis and Analysis of Repeats in Selected Mt Genome Sequences from RefSeq
3. Results
3.1. M. branickii Mt Genome Assemblies and Features
3.2. Non-Coding Regions of the Large Mt Genomes of Baikalian Amphipods
3.3. Mt Gene Order of M. branickii and Its Phylogenetic Position within Baikalian Amphipods
3.4. Statistical Analysis Reveals Genome Length Modes in Invertebrate Phyla
3.5. Phylogenetic Analysis and Repeat Pattern Analysis of the Long Mt Genomes Sequences of Invertebrates
4. Discussion
4.1. The Unusual Architecture of Mt Genome of M. branickii and Its Potential Usefulness for Studies of Mt Genome Transcription and its Regulation
4.2. Features of the Long Mt Genomes in Invertebrates and Putative Mechanisms of Mt Genome Lengthening
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, D.R. The past, present and future of mitochondrial genomics: Have we sequenced enough mtDNAs? Brief. Funct. Genom. 2016, 15, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Vasileiou, P.V.S.; Mourouzis, I.; Pantos, C. Principal aspects regarding the maintenance of mammalian mitochondrial genome integrity. Int. J. Mol. Sci. 2017, 18, 1821. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Pett, W. Animal mitochondrial DNA as we do not know it: Mt-genome organization and evolution in nonbilaterian lineages. Genome Biol. Evol. 2016, 8, 2896–2913. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef]
- Breton, S.; Milani, L.; Ghiselli, F.; Guerra, D.; Stewart, D.T.; Passamonti, M. A resourceful genome: Updating the functional repertoire and evolutionary role of animal mitochondrial DNAs. Trends Genet. 2014, 30, 555–564. [Google Scholar] [CrossRef]
- Ladoukakis, E.D.; Zouros, E. Evolution and inheritance of animal mitochondrial DNA: Rules and exceptions. J. Biol. Res. Thessaloniki 2017, 24, 2. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Graf, S.; Chaga, O.Y.; Lavrov, D.V. Mitochondrial genome of the moon jelly Aurelia aurita (Cnidaria, Scyphozoa): A linear DNA molecule encoding a putative DNA-dependent DNA polymerase. Gene 2006, 381, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Pan, H.C.; Fang, H.Y.; Li, S.W.; Liu, J.H.; Wang, Y.; Wang, A.T. The complete mitochondrial genome of Hydra vulgaris (Hydroida: Hydridae). Mitochondrial DNA 2014, 25, 418–419. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Pett, W.; Voigt, O.; Wörheide, G.; Forget, L.; Lang, B.F.; Kayal, E. Mitochondrial DNA of Clathrina clathrus (Calcarea, Calcinea): Six linear chromosomes, fragmented rRNAs, tRNA editing, and a novel genetic code. Mol. Biol. Evol. 2013, 30, 865–880. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Kern, E.; Park, C.; Nadler, S.A.; Bae, Y.J.; Park, J.K. The bipartite mitochondrial genome of Ruizia karukerae (Rhigonematomorpha, Nematoda). Sci. Rep. 2018, 8, 7482. [Google Scholar] [CrossRef] [PubMed]
- Song, F.; Li, H.; Liu, G.H.; Wang, W.; James, P.; Colwell, D.D.; Tran, A.; Gong, S.; Cai, W.; Shao, R. Mitochondrial genome fragmentation unites the parasitic lice of Eutherian mammals. Syst. Biol. 2019, 68, 430–440. [Google Scholar] [CrossRef]
- Suga, K.; Mark Welch, D.B.; Tanaka, Y.; Sakakura, Y.; Hagiwara, A. Two circular chromosomes of unequal copy number make up the mitochondrial genome of the rotifer Brachionus plicatilis. Mol. Biol. Evol. 2008, 25, 1129–1137. [Google Scholar] [CrossRef]
- Jühling, F.; Pütz, J.; Bernt, M.; Donath, A.; Middendorf, M.; Florentz, C.; Stadler, P.F. Improved systematic tRNA gene annotation allows new insights into the evolution of mitochondrial tRNA structures and into the mechanisms of mitochondrial genome rearrangements. Nucleic Acids Res. 2012, 40, 2833–2845. [Google Scholar] [CrossRef] [PubMed]
- Schirtzinger, E.E.; Tavares, E.S.; Gonzales, L.A.; Eberhard, J.R.; Miyaki, C.Y.; Sanchez, J.J.; Hernandez, A.; Müeller, H.; Graves, G.R.; Fleischer, R.C.; et al. Multiple independent origins of mitochondrial control region duplications in the order Psittaciformes. Mol. Phylogenet. Evol. 2012, 64, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.T.; Foster, P.G.; Hughes, C.; Harper, E.M.; Taylor, J.D.; Littlewood, D.T.J.; Dyal, P.; Hopkins, K.P.; Briscoe, A.G. Curious bivalves: Systematic utility and unusual properties of anomalodesmatan mitochondrial genomes. Mol. Phylogenet. Evol. 2017, 110, 60–72. [Google Scholar] [CrossRef] [PubMed]
- Passamonti, M.; Ricci, A.; Milani, L.; Ghiselli, F. Mitochondrial genomes and Doubly Uniparental Inheritance: New insights from Musculista senhousia sex-linked mitochondrial DNAs (Bivalvia Mytilidae). BMC Genom. 2011, 12, 442. [Google Scholar] [CrossRef] [PubMed]
- Yokobori, S.I.; Fukuda, N.; Nakamura, M.; Aoyama, T.; Oshima, T. Long-term conservation of six duplicated structural genes in cephalopod mitochondrial genomes. Mol. Biol. Evol. 2004, 21, 2034–2046. [Google Scholar] [CrossRef]
- Azevedo, J.L.; Hyman, B.C. Molecular characterization of lengthy mitochondrial DNA duplications from the parasitic nematode Romanomermis culicivorax. Genetics 1993, 133, 933–942. [Google Scholar] [CrossRef]
- Kong, L.; Li, Y.; Kocot, K.M.; Yang, Y.; Qi, L.; Li, Q.; Halanych, K.M. Mitogenomics reveals phylogenetic relationships of Arcoida (Mollusca, Bivalvia) and multiple independent expansions and contractions in mitochondrial genome size. Mol. Phylogenet. Evol. 2020, 150, 106857. [Google Scholar] [CrossRef]
- Liu, Y.G.; Kurokawa, T.; Sekino, M.; Tanabe, T.; Watanabe, K. Complete mitochondrial DNA sequence of the ark shell Scapharca broughtonii: An ultra-large metazoan mitochondrial genome. Comp. Biochem. Physiol.-D Genom. Proteom. 2013, 8, 72–81. [Google Scholar] [CrossRef]
- Lavrov, D.V.; Maikova, O.O.; Pett, W.; Belikov, S.I. Small inverted repeats drive mitochondrial genome evolution in Lake Baikal sponges. Gene 2012, 505, 91–99. [Google Scholar] [CrossRef]
- Romanova, E.V.; Bukin, Y.S.; Mikhailov, K.V.; Logacheva, M.D.; Aleoshin, V.V.; Sherbakov, D.Y. Hidden cases of tRNA gene duplication and remolding in mitochondrial genomes of amphipods. Mol. Phylogenet. Evol. 2020, 144, 106710. [Google Scholar] [CrossRef] [PubMed]
- Chong, R.A.; Mueller, R.L. Polymorphic duplicate genes and persistent non-coding sequences reveal heterogeneous patterns of mitochondrial DNA loss in salamanders. BMC Genom. 2017, 18, 992. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.; Li, B.; Ma, X.; Xu, Y. Evolutionary progression of mitochondrial gene rearrangements and phylogenetic relationships in Strigidae (Strigiformes). Gene 2018, 674, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, Y.; Liu, N.; Yang, J.; Lei, F. Seven complete mitochondrial genome sequences of bushtits (Passeriformes, Aegithalidae, Aegithalos): The evolution pattern in duplicated control regions. Mitochondrial DNA 2015, 26, 350–356. [Google Scholar] [CrossRef]
- Eberhard, J.R.; Wright, T.F. Rearrangement and evolution of mitochondrial genomes in parrots. Mol. Phylogenet. Evol. 2016, 94, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Sayadi, A.; Immonen, E.; Tellgren-Roth, C.; Arnqvist, G. The evolution of dark matter in the mitogenome of seed beetles. Genome Biol. Evol. 2017, 9, 2697–2706. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V.; Forget, L.; Kelly, M.; Lang, B.F. Mitochondrial genomes of two demosponges provide insights into an early stage of animal evolution. Mol. Biol. Evol. 2005, 22, 1231–1239. [Google Scholar] [CrossRef] [PubMed]
- Lukić-Bilela, L.; Brandt, D.; Pojskić, N.; Wiens, M.; Gamulin, V.; Müller, W.E.G. Mitochondrial genome of Suberites domuncula: Palindromes and inverted repeats are abundant in non-coding regions. Gene 2008, 412, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lavrov, D.V. Seventeen new complete mtDNA sequences reveal extensive mitochondrial genome evolution within the Demospongiae. PLoS ONE 2008, 3, e2723. [Google Scholar] [CrossRef] [PubMed]
- Kayal, E.; Bentlage, B.; Collins, A.G.; Kayal, M.; Pirro, S.; Lavrov, D.V. Evolution of linear mitochondrial genomes in medusozoan cnidarians. Genome Biol. Evol. 2012, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, H.; Osigus, H.J.; Rolfes, S.; Kamm, K.; Schierwater, B.; Nakano, H. Mitochondrial genome evolution of placozoans: Gene rearrangements and repeat expansions. Genome Biol. Evol. 2021, 13, evaa213. [Google Scholar] [CrossRef]
- Pont-Kingdon, G.A.; Okada, N.A.; Macfarlane, J.L.; Beagley, C.T.; Wolstenholme, D.R.; Cavalier-Smith, T.; Clark-Walker, G.D. A coral mitochondrial mutS gene. Nature 1995, 375, 109–111. [Google Scholar] [CrossRef]
- Bilewitch, J.P.; Degnan, S.M. A unique horizontal gene transfer event has provided the octocoral mitochondrial genome with an active mismatch repair gene that has potential for an unusual self-contained function. BMC Evol. Biol. 2011, 11, 228. [Google Scholar] [CrossRef] [PubMed]
- Shimpi, G.G.; Vargas, S.; Poliseno, A.; Wörheide, G. Mitochondrial RNA processing in absence of tRNA punctuations in octocorals. BMC Mol. Biol. 2017, 18, 16. [Google Scholar] [CrossRef]
- Szitenberg, A.; Rot, C.; Ilan, M.; Huchon, D. Diversity of sponge mitochondrial introns revealed by cox 1 sequences of Tetillidae. BMC Evol. Biol. 2010, 10, 288. [Google Scholar] [CrossRef] [PubMed]
- Banguera-Hinestroza, E.; Ferrada, E.; Sawall, Y.; Flot, J.F. Computational Characterization of the mtORF of Pocilloporid Corals: Insights into Protein Structure and Function in Stylophora Lineages from Contrasting Environments. Genes 2019, 10, 324. [Google Scholar] [CrossRef] [PubMed]
- Chi, S.I.; Urbarova, I.; Johansen, S.D. Expression of homing endonuclease gene and insertion-like element in sea anemone mitochondrial genomes: Lesson learned from Anemonia viridis. Gene 2018, 652, 78–86. [Google Scholar] [CrossRef]
- Szafranski, P. Evolutionarily recent, insertional fission of mitochondrial cox2 into complementary genes in bilaterian Metazoa. BMC Genom. 2017, 18, 269. [Google Scholar] [CrossRef]
- Breton, S.; Stewart, D.T.; Shepardson, S.; Trdan, R.J.; Bogan, A.E.; Chapman, E.G.; Ruminas, A.J.; Piontkivska, H.; Hoeh, W.R. Novel protein genes in animal mtDNA: A new sex determination system in freshwater mussels (Bivalvia: Unionoida)? Mol. Biol. Evol. 2011, 28, 1645–1659. [Google Scholar] [CrossRef]
- Passamonti, M.; Calderone, M.; Delpero, M.; Plazzi, F. Clues of in vivo nuclear gene regulation by mitochondrial short non-coding RNAs. Sci. Rep. 2020, 10, 8219. [Google Scholar] [CrossRef] [PubMed]
- Plaza, S.; Menschaert, G.; Payre, F. In search of lost small peptides. Annu. Rev. Cell Dev. Biol. 2017, 33, 391–416. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, A.; Plazzi, F.; Milani, L.; Ghiselli, F.; Passamonti, M. SmithRNAs: Could mitochondria “bend” nuclear regulation? Mol. Biol. Evol. 2017, 34, 1960–1973. [Google Scholar] [CrossRef] [PubMed]
- Aguado, M.T.; Grande, C.; Gerth, M.; Bleidorn, C.; Noreña, C. Characterization of the complete mitochondrial genomes from Polycladida (Platyhelminthes) using next-generation sequencing. Gene 2016, 575, 199–205. [Google Scholar] [CrossRef]
- Zhang, D.; Li, W.X.; Zou, H.; Wu, S.G.; Li, M.; Jakovlić, I.; Zhang, J.; Chen, R.; Wang, G.T. Mitochondrial genomes of two diplectanids (Platyhelminthes: Monogenea) expose paraphyly of the order Dactylogyridea and extensive tRNA gene rearrangements. Parasites Vectors 2018, 11, 601. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, X.; Yu, Z.; Wei, Z.; Xia, J. Comparison of seven Crassostrea mitogenomes and phylogenetic analyses. Mol. Phylogenet. Evol. 2010, 57, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Palomares-Rius, J.E.; Cantalapiedra-Navarrete, C.; Archidona-Yuste, A.; Blok, V.C.; Castillo, P. Mitochondrial genome diversity in dagger and needle nematodes (Nematoda: Longidoridae). Sci. Rep. 2017, 7, 41813. [Google Scholar] [CrossRef] [PubMed]
- Rosengarten, R.D.; Sperling, E.A.; Moreno, M.A.; Leys, S.P.; Dellaporta, S.L. The mitochondrial genome of the hexactinellid sponge Aphrocallistes vastus: Evidence for programmed translational frameshifting. BMC Genom. 2008, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, A.; Feregrino, C.; Sartoretto, S.; Aurelle, D.; Wörheide, G.; McFadden, C.S.; Vargas, S. Comparative mitogenomics, phylogeny and evolutionary history of Leptogorgia (Gorgoniidae). Mol. Phylogenet. Evol. 2017, 115, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Helfenbein, K.G.; Fourcade, H.M.; Vanjani, R.G.; Boore, J.L. The mitochondrial genome of Paraspadella gotoi is highly reduced and reveals that chaetognaths are a sister group to protostomes. Proc. Natl. Acad. Sci. USA 2004, 101, 10639–10643. [Google Scholar] [CrossRef]
- Miyamoto, H.; Machida, R.J.; Nishida, S. Complete mitochondrial genome sequences of the three pelagic chaetognaths Sagitta nagae, Sagitta decipiens and Sagitta enflata. Comp. Biochem. Physiol.-D Genom. Proteom. 2010, 5, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Arafat, H.; Alamaru, A.; Gissi, C.; Huchon, D. Extensive mitochondrial gene rearrangements in Ctenophora: Insights from benthic Platyctenida. BMC Evol. Biol. 2018, 18, 65. [Google Scholar] [CrossRef] [PubMed]
- Schultz, D.T.; Eizenga, J.M.; Corbett-Detig, R.B.; Francis, W.R.; Christianson, L.M.; Haddock, S.H. Conserved novel ORFs in the mitochondrial genome of the ctenophore Beroe forskalii. PeerJ 2020, 8, e8356. [Google Scholar] [CrossRef] [PubMed]
- Pett, W.; Ryan, J.F.; Pang, K.; Mullikin, J.C.; Martindale, M.Q.; Baxevanis, A.D.; Lavrov, D.V. Extreme mitochondrial evolution in the ctenophore Mnemiopsis leidyi: Insight from mtDNA and the nuclear genome. Mitochondrial DNA 2011, 22, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Barthélémy, R.M.; Seligmann, H. Cryptic tRNAs in chaetognath mitochondrial genomes. Comput. Biol. Chem. 2016, 62, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Dermauw, W.; Vanholme, B.; Tirry, L.; Van Leeuwen, T. Mitochondrial genome analysis of the predatory mite Phytoseiulus persimilis and a revisit of the Metaseiulus occidentalis mitochondrial genome. Genome 2010, 53, 285–301. [Google Scholar] [CrossRef] [PubMed]
- Egger, B.; Bachmann, L.; Fromm, B. Atp8 is in the ground pattern of flatworm mitochondrial genomes. BMC Genom. 2017, 18, 414. [Google Scholar] [CrossRef] [PubMed]
- Gan, H.M.; Grandjean, F.; Jenkins, T.L.; Austin, C.M. Absence of evidence is not evidence of absence: Nanopore sequencing and complete assembly of the European lobster (Homarus gammarus) mitogenome uncovers the missing nad2 and a new major gene cluster duplication. BMC Genom. 2019, 20, 335. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, S.; Li, Y.; Liu, B. Complete mtDNA of Meretrix lusoria (Bivalvia: Veneridae) reveals the presence of an atp8 gene, length variation and heteroplasmy in the control region. Comp. Biochem. Physiol.-D Genom. Proteom. 2010, 5, 256–264. [Google Scholar] [CrossRef]
- Pu, L.; Liu, H.; Wang, G.; Li, B.; Xia, G.; Shen, M.; Yang, M. Complete mitochondrial genome of the cockle Anadara antiquata (Linnaeus, 1758). Mitochondrial DNA Part B 2019, 4, 2293–2294. [Google Scholar] [CrossRef]
- Osigus, H.J.; Rolfes, S.; Herzog, R.; Kamm, K.; Schierwater, B. Polyplacotoma mediterranea is a new ramified placozoan species. Curr. Biol. 2019, 29, R148–R149. [Google Scholar] [CrossRef] [PubMed]
- Signorovitch, A.Y.; Buss, L.W.; Dellaporta, S.L. Comparative genomics of large mitochondria in placozoans. PLoS Genet. 2007, 3, e13. [Google Scholar] [CrossRef] [PubMed]
- Lavrov, D.V. Rapid proliferation of repetitive palindromic elements in mtDNA of the endemic Baikalian sponge Lubomirskia baicalensis. Mol. Biol. Evol. 2010, 27, 757–760. [Google Scholar] [CrossRef]
- Ohta, T. Slightly deleterious mutant substitutions in evolution. Nature 1973, 246, 96–98. [Google Scholar] [CrossRef]
- Ghiselli, F.; Milani, L.; Guerra, D.; Chang, P.L.; Breton, S.; Nuzhdin, S.V.; Passamonti, M. Structure, transcription, and variability of metazoan mitochondrial genome: Perspectives from an unusual mitochondrial inheritance system. Genome Biol. Evol. 2013, 5, 1535–1554. [Google Scholar] [CrossRef] [PubMed]
- Klucnika, A.; Ma, H. A battle for transmission: The cooperative and selfish animal mitochondrial genomes. Open Biol. J. 2019, 9, 180267. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.; Koskella, B.; Schaack, S. Mutation pressure and the evolution of organelle genomic architecture. Science 2006, 311, 1727–1730. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L. The duplication/random loss model for gene rearrangement exemplified by mitochondrial genomes of deuterostome animals. In Comparative Genomics, 1st ed.; Sankoff, D., Nadeau, J.H., Eds.; Kluwer Academic: Boston, UK, 2000; Volume 1, pp. 133–147. [Google Scholar]
- Levinson, G.; Gutman, G.A. Slipped-strand mispairing: A major mechanism for DNA sequence evolution. Mol. Biol. Evol. 1987, 4, 203–221. [Google Scholar]
- Mjelle, K.A.; Karlsen, B.O.; Jorgensen, T.E.; Moum, T.; Johansen, S.D. Halibut mitochondrial genomes contain extensive heteroplasmic tandem repeat arrays involved in DNA recombination. BMC Genom. 2008, 9, 10. [Google Scholar] [CrossRef] [PubMed]
- Nardi, F.; Carapelli, A.; Frati, F. Repeated regions in mitochondrial genomes: Distribution, origin and evolutionary significance. Mitochondrion 2012, 12, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.R.; Lee, R.W. The mitochondrial and plastid genomes of Volvox carteri: Bloated molecules rich in repetitive DNA. BMC Genom. 2009, 10, 132. [Google Scholar] [CrossRef] [PubMed]
- Selosse, M.A.; Albert, B.; Godelle, B. Reducing the genome size of organelles favours gene transfer to the nucleus. Trends Ecol. Evol. 2001, 16, 135–141. [Google Scholar] [CrossRef]
- Rand, D.M. Endotherms, ectotherms, and mitochondrial genome-size variation. J. Mol. Evol. 1993, 37, 281–295. [Google Scholar] [CrossRef] [PubMed]
- Rand, D.M. Population genetics of the cytoplasm and the units of selection on mitochondrial DNA in Drosophila melanogaster. Genetica 2011, 139, 685–697. [Google Scholar] [CrossRef][Green Version]
- Ma, H.; O’Farrell, P.H. Selfish drive can trump function when animal mitochondrial genomes compete. Nat. Genet. 2016, 48, 798–802. [Google Scholar] [CrossRef]
- Gusic, M.; Prokisch, H. ncRNAs: New players in mitochondrial health and disease? Front. Genet. 2020, 11, 95. [Google Scholar] [CrossRef]
- Reynolds, J.C.; Bwiza, C.P.; Lee, C. Mitonuclear genomics and aging. Hum. Genet. 2020, 139, 381–399. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.; Ma, H.Y.; Park, C.; Ortogero, N.; Song, R.; Hennig, G.W.; Zheng, H.; Lin, Y.M.; Moro, L.; Hsieh, J.T.; et al. The mitochondrial genome encodes abundant small noncoding RNAs. Cell Res. 2013, 23, 759–774. [Google Scholar] [CrossRef]
- Capt, C.; Passamonti, M.; Breton, S. The human mitochondrial genome may code for more than 13 proteins. Mitochondrial DNA 2016, 27, 3098–3101. [Google Scholar] [CrossRef]
- Romanova, E.V.; Aleoshin, V.V.; Kamaltynov, R.M.; Mikhailov, K.V.; Logacheva, M.D.; Sirotinina, E.A.; Gornov, A.Y.; Anikin, A.S.; Sherbakov, D.Y. Evolution of mitochondrial genomes in Baikalian amphipods. BMC Genom. 2016, 17, 1016. [Google Scholar] [CrossRef] [PubMed]
- Bazikalova, A.Y. Lake Baikal amphipods. In Proceedings of the Baikal Limnological Station; Vereshagin, G.Y., Ed.; Academy of Sciences Publishing House: Moscow, Russia, 1945; pp. 1–440. [Google Scholar]
- Kamaltynov, R.M. Amfipoda: Gammaroidea in Angara and Yenisei rivers. In Index of Animal Species Inhabiting Lake Baikal and Its Catchment Area; Timoshkin, O.A., Proviz, V.I., Sitnikova, T.Y., Slugina, Z.V., Melnik, N.G., Eds.; Nauka: Novosibirsk, Russia, 2009; Volume 2, pp. 297–329. [Google Scholar]
- Bekman, M.Y.; Afanasyeva, E.L. Distribution and production of Macrohectopus in Lake Baikal. In Proceedings of Limnological Institute; Biological Productivity of Baikal Pelagic Region and Its Dynamics; Nauka: Novosibirsk, Russia, 1977; Volume 19, pp. 76–98. [Google Scholar]
- Naumova, E.Y.; Zaidykov, I.Y.; Makarov, M.M. Recent quantitative values of Macrohectopus branickii (Dyb.) (amphipoda) from Lake Baikal. J. Gt. Lakes Res. 2020, 46, 48–52. [Google Scholar] [CrossRef]
- Karnaukhov, D.Y.; Bedulina, D.S.; Kaus, A.; Prokosov, S.O.; Sartoris, L.; Timofeyev, M.A.; Takhteev, V.V. Behaviour of Lake Baikal amphipods as a part of the night migratory complex in the Kluevka settlement region (South-Eastern Baikal). Crustaceana 2016, 89, 419–430. [Google Scholar] [CrossRef]
- Takhteev, V.V.; Karnaukhov, D.Y.; Govorukhina, E.B.; Misharin, A.S. Diel vertical migrations of hydrobionts in the coastal area of Lake Baikal. Inland Water Biol. 2019, 12, 178–189. [Google Scholar] [CrossRef]
- Rudstam, L.G.; Melnik, N.G.; Timoshkin, O.A.; Hansson, S.; Pushkin, S.V.; Nemov, V. Diel dynamics of an aggregation of Macrohectopus branickii (Dyb.) (Amphipoda, Gammaridae) in the Barguzin Bay, Lake Baikal, Russia. J. Gt. Lakes Res. 1992, 18, 286–297. [Google Scholar] [CrossRef]
- Doyle, J.J.; Dickson, E.E. Preservation of plant samples for DNA restriction endonuclease analysis. Taxon 1987, 36, 715–722. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Naumenko, S.A.; Logacheva, M.D.; Popova, N.V.; Klepikova, A.V.; Penin, A.A.; Bazykin, G.A.; Etingova, A.E.; Mugue, N.S.; Kondrashov, A.S.; Yampolsky, L.Y. Transcriptome-based phylogeny of endemic Lake Baikal amphipod species flock: Fast speciation accompanied by frequent episodes of positive selection. Mol. Ecol. 2017, 26, 536–553. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kerpedjiev, P.; Hammer, S.; Hofacker, I.L. Forna (force-directed RNA): Simple and effective online RNA secondary structure diagrams. Bioinformatics 2015, 31, 3377–3379. [Google Scholar] [CrossRef] [PubMed]
- Greiner, S.; Lehwark, P.; Bock, R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 2019, 47, W59–W64. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Zardoya, R.; Telford, M.J. TranslatorX: Multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 2010, 38, W7–W13. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; Von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Boil. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree-v1.4.3. Available online: http://tree.bio.ed.ac.uk/software/figtree (accessed on 12 March 2021).
- Kassambara, A. Machine Learning Essentials: Practical Guide in R. STHDA. Available online: http://www.sthda.com/english/articles/38-regression-model-validation/158-regression-model-accuracy-metrics-r-square-aic-bic-cp-and-more/ (accessed on 18 November 2020).
- Basso, D.; Pesarin, F.; Salmaso, L.; Solari, A. Synchronized Permutation Tests in Two-way ANOVA. In Permutation Tests for Stochastic Ordering and ANOVA; Lecture Notes in Statistics; Springer: New York, NY, USA, 2009; Volume 3. [Google Scholar]
- Xia, X. DAMBE6: New tools for microbial genomics, phylogenetics, and molecular evolution. J. Hered. 2017, 10, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Bekman, M.Y. Ecology and production of Micruropus possolskii sow. and Gmelinoides fasciatus stebb. In System and Ecology of Crustaceans of Lake Baikal; Galaziy, G.I., Ed.; Academy of Sciences Publishing House: Moscow, Russia, 1962; pp. 141–156. [Google Scholar]
- Timofeyev, M.A.; Shatilina, J.M.; Stom, D.I. Attitude to temperature factor of some endemic amphipods from Lake Baikal and Holarctic Gammarus lacustris Sars, 1863: A comparative experimental study. Arthropoda Sel. 2001, 10, 93. [Google Scholar]
- Macdonald, K.S., III; Yampolsky, L.; Duffy, J.E. Molecular and morphological evolution of the amphipod radiation of Lake Baikal. Mol. Phylogenet. Evol. 2005, 35, 323–343. [Google Scholar] [CrossRef]
- Smith, D.R.; Snyder, M. Complete mitochondrial DNA sequence of the scallop Placopecten magellanicus: Evidence of transposition leading to an uncharacteristically large mitochondrial genome. J. Mol. Evol. 2007, 65, 380–391. [Google Scholar] [CrossRef]
- Chapman, E.G.; Piontkivska, H.; Walker, J.M.; Stewart, D.T.; Curole, J.P.; Hoeh, W.R. Extreme primary and secondary protein structure variability in the chimeric male-transmitted cytochrome c oxidase subunit II protein in freshwater mussels: Evidence for an elevated amino acid substitution rate in the face of domain-specific purifying selection. BMC Evol. Biol. 2008, 8, 165. [Google Scholar]
- Stöger, I.; Schrödl, M. Mitogenomics does not resolve deep molluscan relationships (yet?). Mol. Phylogenet. Evol. 2013, 69, 376–392. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Walker, J.M.; Stewart, D.T.; Trdan, R.J.; Vijayaraghavan, S.; Curole, J.P.; Hoeh, W.R. Presence of a unique male-specific extension of C-terminus to the cytochrome c oxidase subunit II protein coded by the male-transmitted mitochondrial genome of Venustaconcha ellipsiformis (Bivalvia: Unionoidea). FEBS Lett. 2006, 580, 862–866. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ren, X.; Bi, Y.; Ding, Q.; Ho, V.W.S.; Zhao, Z. Comparative mitochondrial genomics reveals a possible role of a recent duplication of NADH dehydrogenase subunit 5 in gene regulation. DNA Res. 2018, 25, 577–586. [Google Scholar] [CrossRef]
- Dietrich, A.; Wallet, C.; Iqbal, R.K.; Gualberto, J.M.; Lotfi, F. Organellar non-coding RNAs: Emerging regulation mechanisms. Biochimie 2015, 117, 48–62. [Google Scholar] [CrossRef] [PubMed]
- Milligan, M.J.; Harvey, E.; Yu, A.; Morgan, A.L.; Smith, D.L.; Zhang, E.; Berengut, J.; Sivananthan, J.; Subramaniam, R.; Skoric, A.; et al. Global intersection of long non-coding RNAs with processed and unprocessed pseudogenes in the human genome. Front. Genet. 2016, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA transcription and its regulation: An evolutionary perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef]
- De Lay, N.R.; Garsin, D.A. The unmasking of ‘junk’RNA reveals novel sRNAs: From processed RNA fragments to marooned riboswitches. Curr. Opin. Microbiol. 2016, 30, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Schimmel, P. The emerging complexity of the tRNA world: Mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 2018, 19, 45–58. [Google Scholar] [CrossRef]
- Lavrov, D.V. Key transitions in animal evolution: A mitochondrial DNA perspective. Integr. Comp. Biol. 2007, 47, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Pleše, B.; Lukić-Bilela, L.; Bruvo-Mađarić, B.; Harcet, M.; Imešek, M.; Bilandžija, H.; Ćetković, H. The mitochondrial genome of stygobitic sponge Eunapius subterraneus: mtDNA is highly conserved in freshwater sponges. Hydrobiologia 2012, 687, 49–59. [Google Scholar] [CrossRef]
- Schuster, A.; Vargas, S.; Knapp, I.S.; Pomponi, S.A.; Toonen, R.J.; Erpenbeck, D.; Wörheide, G. Divergence times in demosponges (Porifera): First insights from new mitogenomes and the inclusion of fossils in a birth-death clock model. BMC Evol. Biol. 2018, 18, 114. [Google Scholar] [CrossRef] [PubMed]
- Peretolchina, T.E.; Sitnikova, T.Y.; Sherbakov, D.Y. The complete mitochondrial genomes of four Baikal molluscs from the endemic family Baicaliidae (Caenogastropoda: Truncatelloida). J. Molluscan Stud. 2020, 86, 201–209. [Google Scholar] [CrossRef]
- Mats, V.D.; Shcherbakov, D.Y.; Efimova, I.M. Late Cretaceous-Cenozoic history of the Lake Baikal depression and formation of its unique biodiversity. Stratigr. Geol. Correl. 2011, 19, 404–423. [Google Scholar] [CrossRef]
- Zubakov, D.Y.; Sherbakov, D.Y.; Sitnikova, T.Y. Analysis of phylogenetic relationships of endemic baikalian mollusks Baicaliidae family based on nucleotide sequences partial mitochondrial CO1 gene. Mol. Biol. 1997, 31, 32–36. (In Russian) [Google Scholar]
- Chen, Y.J.; Kim, S.; Wan, X. Mitochondrial genomes of the Dorcus velutinus complex (Coleoptera: Lucanidae) with the large intergenic spacer showing unique short sequence repeats and their implications for systematics. J. Asia Pac. Entomol. 2021, 24, 493–501. [Google Scholar] [CrossRef]
- Mohandas, N.; Pozio, E.; La Rosa, G.; Korhonen, P.K.; Young, N.D.; Koehler, A.V.; Hall, R.S.; Sternberg, P.W.; Boag, P.R.; Jex, A.R.; et al. Mitochondrial genomes of Trichinella species and genotypes–A basis for diagnosis, and systematic and epidemiological explorations. Int. J. Parasitol. 2014, 44, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.F.; Xu, S.X.; Zhu, C.C.; Jia, X.N.; Zhou, X.M.; Zou, J.X. The complete mitochondrial genome of the freshwater crab Longpotamon kenliense (Decapoda, Brachyura, Potamidae) with phylogenetic consideration. Crustaceana 2020, 93, 1277–1293. [Google Scholar] [CrossRef]
- Feng, Y.; Li, Q.; Yu, H.; Kong, L. Complete mitochondrial genome sequence of Cucullaea labiata (Arcoida: Cucullaeidae) and phylogenetic implications. Genes Genom. 2017, 39, 867–875. [Google Scholar] [CrossRef]
- Xu, K.; Kanno, M.; Yu, H.; Li, Q.; Kijima, A. Complete mitochondrial DNA sequence and phylogenetic analysis of Zhikong scallop Chlamys farreri (Bivalvia: Pectinidae). Mol. Biol. Rep. 2011, 38, 3067–3074. [Google Scholar] [CrossRef]
- Sun, L.; Zhuo, K.; Wang, H.; Song, H.; Chi, W.; Zhang, L.H.; Liao, J. The complete mitochondrial genome of Aphelenchoides besseyi (Nematoda: Aphelenchoididae), the first sequenced representative of the subfamily Aphelenchoidinae. Nematology 2014, 16, 1167–1180. [Google Scholar] [CrossRef]
- Li, Y.; Kocot, K.M.; Schander, C.; Santos, S.R.; Thornhill, D.J.; Halanych, K.M. Mitogenomics reveals phylogeny and repeated motifs in control regions of the deep-sea family Siboglinidae (Annelida). Mol. Phylogenet. Evol. 2015, 85, 221–229. [Google Scholar] [CrossRef]
- Seixas, V.C.; de Moraes Russo, C.A.; Paiva, P.C. Mitochondrial genome of the Christmas tree worm Spirobranchus giganteus (Annelida: Serpulidae) reveals a high substitution rate among annelids. Gene 2017, 605, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.E.; Lapraz, F.; Egger, B.; Telford, M.J.; Schiffer, P.H. The mitochondrial genomes of the acoelomorph worms Paratomella rubra, Isodiametra pulchra and Archaphanostoma ylvae. Sci. Rep. 2017, 7, 1847. [Google Scholar] [CrossRef] [PubMed]
- Korkmaz, E.M.; Budak, M.; Ördek, M.N.; Başıbüyük, H.H. The complete mitogenomes of Calameuta filiformis (Eversmann, 1847) and Calameuta idolon (Rossi, 1794) (Hymenoptera: Cephidae): The remarkable features of the elongated A+ T rich region in Cephini. Gene 2016, 576, 404–411. [Google Scholar] [CrossRef]
- Humphreys-Pereira, D.A.; Elling, A.A. Mitochondrial genomes of Meloidogyne chitwoodi and M. incognita (Nematoda: Tylenchina): Comparative analysis, gene order and phylogenetic relationships with other nematodes. Mol. Biochem. Parasitol. 2014, 194, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Boore, J.L.; Brown, W.M. Big trees from little genomes: Mitochondrial gene order as a phylogenetic tool. Curr. Opin. Genet. Dev. 1998, 8, 668–674. [Google Scholar] [CrossRef]
- Hyman, B.C.; Lewis, S.C.; Tang, S.; Wu, Z. Rampant gene rearrangement and haplotype hypervariation among nematode mitochondrial genomes. Genetica 2011, 139, 611–615. [Google Scholar] [CrossRef][Green Version]
- Dahal, S.; Dubey, S.; Raghavan, S.C. Homologous recombination-mediated repair of DNA double-strand breaks operates in mammalian mitochondria. Cell. Mol. Life Sci. 2018, 75, 1641–1655. [Google Scholar] [CrossRef]
- Davila, J.I.; Arrieta-Montiel, M.P.; Wamboldt, Y.; Cao, J.; Hagmann, J.; Shedge, V.; Xu, Y.Z.; Weigel, D.; Mackenzie, S.A. Double-strand break repair processes drive evolution of the mitochondrial genome in Arabidopsis. BMC Biol. 2011, 9, 64. [Google Scholar] [CrossRef]
- Tsaousis, A.D.; Martin, D.P.; Ladoukakis, E.D.; Posada, D.; Zouros, E. Widespread recombination in published animal mtDNA sequences. Mol. Biol. Evol. 2005, 22, 925–933. [Google Scholar] [CrossRef][Green Version]
- Fritsch, E.S.; Chabbert, C.D.; Klaus, B.; Steinmetz, L.M. A genome-wide map of mitochondrial DNA recombination in yeast. Genetics 2014, 198, 755–771. [Google Scholar] [CrossRef]
- Kraytsberg, Y.; Schwartz, M.; Brown, T.A.; Ebralidse, K.; Kunz, W.S.; Clayton, D.A.; Vissing, J.; Khrapko, K. Recombination of human mitochondrial DNA. Science 2004, 304, 981. [Google Scholar] [CrossRef] [PubMed]
- Lunt, D.H.; Hyman, B.C. Animal mitochondrial DNA recombination. Nature 1997, 387, 247. [Google Scholar] [CrossRef]
- Erpenbeck, D.; Voigt, O.; Wörheide, G.; Lavrov, D.V. The mitochondrial genomes of sponges provide evidence for multiple invasions by Repetitive Hairpin-forming Elements (RHE). BMC Genom. 2010, 10, 591. [Google Scholar] [CrossRef] [PubMed]
- Bullerwell, C.E.; Leigh, J.; Forget, L.; Lang, B.F. A comparison of three fission yeast mitochondrial genomes. Nucleic Acids Res. 2003, 31, 759–768. [Google Scholar] [CrossRef] [PubMed][Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanova, E.V.; Bukin, Y.S.; Mikhailov, K.V.; Logacheva, M.D.; Aleoshin, V.V.; Sherbakov, D.Y. The Mitochondrial Genome of a Freshwater Pelagic Amphipod Macrohectopus branickii Is among the Longest in Metazoa. Genes 2021, 12, 2030. https://doi.org/10.3390/genes12122030
Romanova EV, Bukin YS, Mikhailov KV, Logacheva MD, Aleoshin VV, Sherbakov DY. The Mitochondrial Genome of a Freshwater Pelagic Amphipod Macrohectopus branickii Is among the Longest in Metazoa. Genes. 2021; 12(12):2030. https://doi.org/10.3390/genes12122030
Chicago/Turabian StyleRomanova, Elena V., Yurij S. Bukin, Kirill V. Mikhailov, Maria D. Logacheva, Vladimir V. Aleoshin, and Dmitry Y. Sherbakov. 2021. "The Mitochondrial Genome of a Freshwater Pelagic Amphipod Macrohectopus branickii Is among the Longest in Metazoa" Genes 12, no. 12: 2030. https://doi.org/10.3390/genes12122030
APA StyleRomanova, E. V., Bukin, Y. S., Mikhailov, K. V., Logacheva, M. D., Aleoshin, V. V., & Sherbakov, D. Y. (2021). The Mitochondrial Genome of a Freshwater Pelagic Amphipod Macrohectopus branickii Is among the Longest in Metazoa. Genes, 12(12), 2030. https://doi.org/10.3390/genes12122030