Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
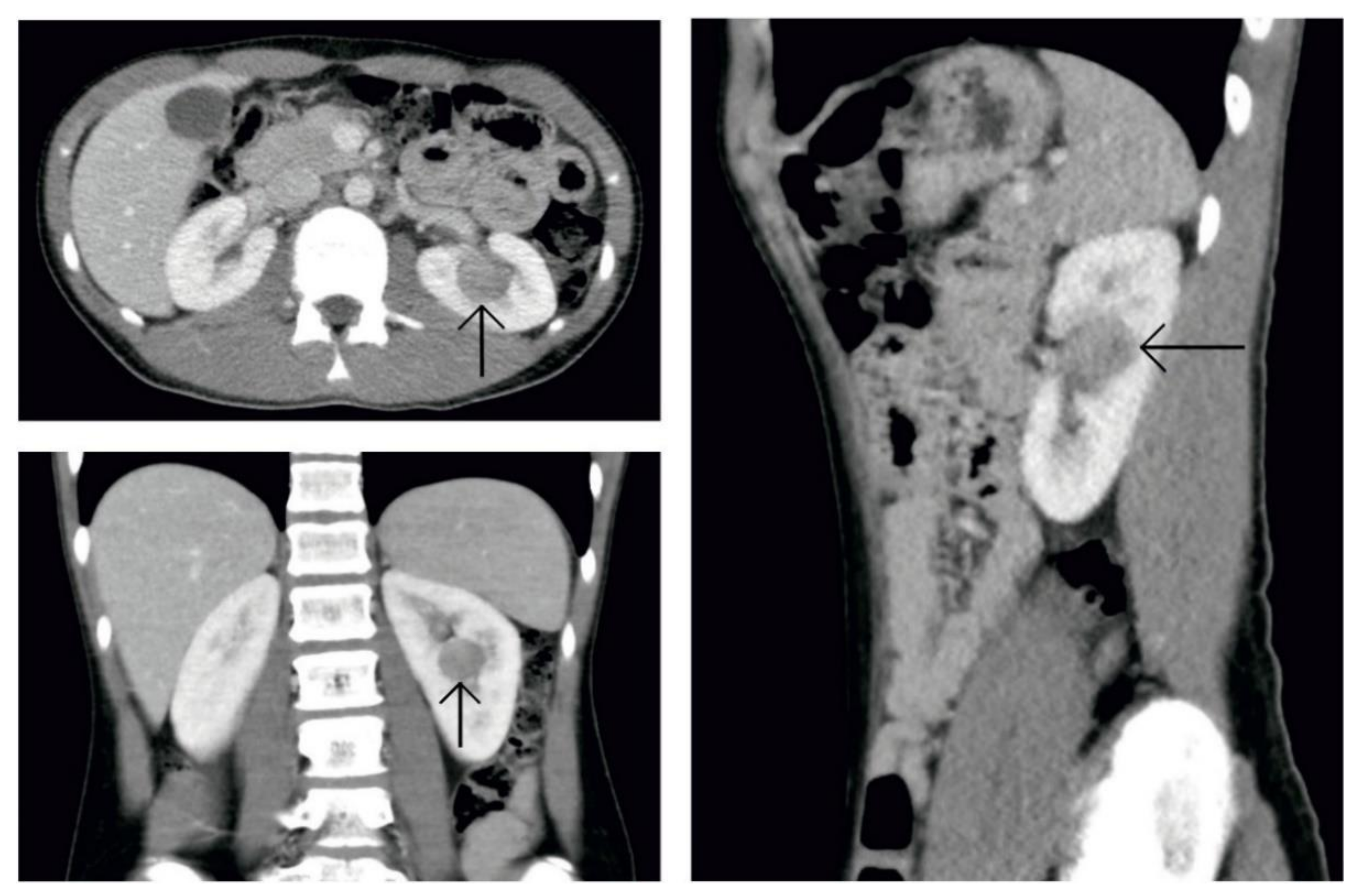
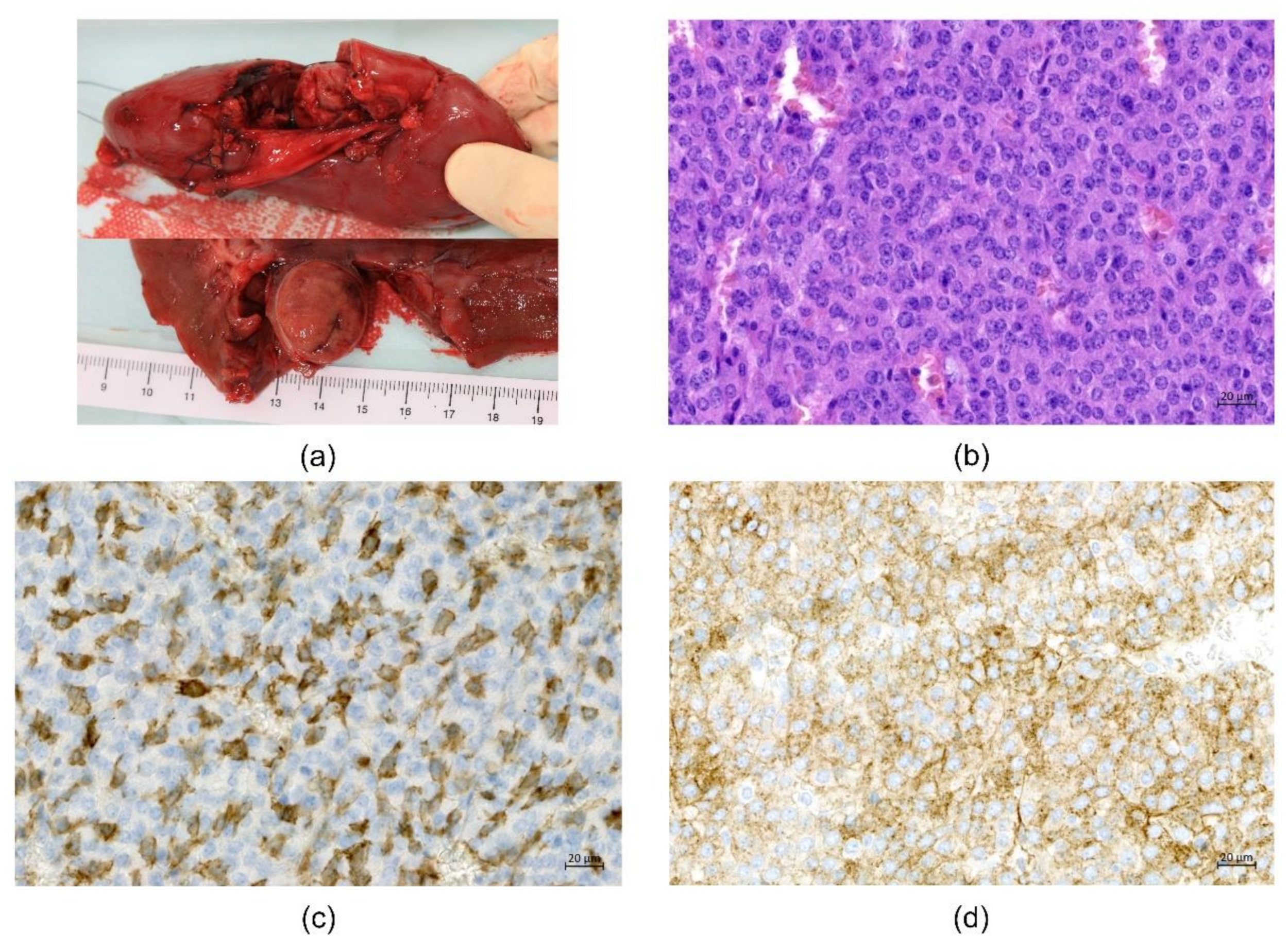
2. Case Presentation
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Requena, L.; Kutzner, H. Hemangioendothelioma. Semin. Diagn. Pathol. 2013, 30, 29–44. [Google Scholar] [CrossRef] [PubMed]
- Pansuriya, T.C.; van Eijk, R.; d’Adamo, P.; van Ruler, M.A.; Kuijjer, M.L.; Oosting, J.; Cleton-Jansen, A.M.; van Oosterwijk, J.G.; Verbeke, S.L.; Meijer, D.; et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat. Genet. 2011, 43, 1256–1261. [Google Scholar] [CrossRef] [PubMed]
- McCrindle, B.W. Assessment and management of hypertension in children and adolescents. Nat. Rev. Cardiol. 2010, 7, 155–163. [Google Scholar] [CrossRef]
- Nunes, I.; Santos, T.; Tavares, J.; Correia, L.; Coutinho, J.; Nogueira, J.M.B.; Carvalho, L.; Soares, J.L.D. Secondary hypertension due to a juxtaglomerular cell tumor. J. Am. Soc. Hypertens. 2018, 12, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Perkins, P.; Weiss, S.W. Spindle cell hemangioendothelioma. An analysis of 78 cases with reassessment of its pathogenesis and biologic behavior. Am. J. Surg. Pathol. 1996, 20, 1196–1204. [Google Scholar] [CrossRef]
- Enjolras, O.; Mulliken, J.; Kozakewich, H. Vascular tumors and tumor-like lesions. In Mulliken & Young’s Vascular Anomalies: Hemangiomas and Malformations, 2nd ed.; Mulliken, J., Burrows, P., Fishman, S., Eds.; Oxford University Press: New York, NY, USA, 2013; pp. 259–324. [Google Scholar]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants:A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Cantor, S.; Drapkin, R.; Zhang, F.; Lin, Y.; Han, J.; Pamidi, S.; Livingston, D.M. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc. Natl. Acad. Sci. USA 2004, 101, 2357–2362. [Google Scholar] [CrossRef] [Green Version]
- Cantor, S.B.; Bell, D.W.; Ganesan, S.; Kass, E.M.; Drapkin, R.; Grossman, S.; Wahrer, D.C.; Sgroi, D.C.; Lane, W.S.; Haber, D.A.; et al. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell 2001, 105, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Kinnersley, B.; Kamatani, Y.; Labussière, M.; Wang, Y.; Galan, P.; Mokhtari, K.; Delattre, J.Y.; Gousias, K.; Schramm, J.; Schoemaker, M.J.; et al. Search for new loci and low-frequency variants influencing glioma risk by exome-array analysis. Eur. J. Hum. Genet. 2016, 24, 717–724. [Google Scholar] [CrossRef] [Green Version]
- Schrader, K.A.; Cheng, D.T.; Joseph, V.; Prasad, M.; Walsh, M.; Zehir, A.; Ni, A.; Thomas, T.; Benayed, R.; Ashraf, A.; et al. Germline Variants in Targeted Tumor Sequencing Using Matched Normal DNA. JAMA Oncol. 2016, 2, 104–111. [Google Scholar] [CrossRef]
- Susswein, L.R.; Marshall, M.L.; Nusbaum, R.; Vogel Postula, K.J.; Weissman, S.M.; Yackowski, L.; Vaccari, E.M.; Bissonnette, J.; Booker, J.K.; Cremona, M.L.; et al. Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genet. Med. 2016, 18, 823–832. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Robertson, P.W.; Klidjian, A.; Harding, L.K.; Walters, G.; Lee, M.R.; Robb-Smith, A.H. Hypertension due to a renin-secreting renal tumour. Am. J. Med. 1967, 43, 963–976. [Google Scholar] [CrossRef]
- Trnka, P.; Orellana, L.; Walsh, M.; Pool, L.; Borzi, P. Reninoma: An uncommon cause of Renin-mediated hypertension. Front. Pediatr. 2014, 2, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osawa, S.; Hosokawa, Y.; Soda, T.; Yasuda, T.; Kaneto, H.; Kitamura, T.; Kozawa, J.; Otsuki, M.; Imagawa, A.; Okumi, M.; et al. Juxtaglomerular cell tumor that was preoperatively diagnosed using selective renal venous sampling. Intern. Med. 2013, 52, 1937–1942. [Google Scholar] [CrossRef] [Green Version]
- Shao, L.; Manalang, M.; Cooley, L. Juxtaglomerular cell tumor in an 8-year-old girl. Pediatr. Blood Cancer 2008, 50, 406–409. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, Z.; Ji, J. Juxtaglomerular cell tumor: A case report. Oncol. Lett. 2016, 11, 1418–1420. [Google Scholar] [CrossRef] [Green Version]
- Wang, F.; Shi, C.; Cui, Y.; Li, C.; Tong, A. Juxtaglomerular cell tumor: Clinical and immunohistochemical features. J. Clin. Hypertens. 2017, 19, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Duan, X.; Bruneval, P.; Hammadeh, R.; Fresco, R.; Eble, J.N.; Clark, J.I.; Vigneswaran, W.T.; Flanigan, R.C.; Picken, M.M. Metastatic juxtaglomerular cell tumor in a 52-year-old man. Am. J. Surg. Pathol. 2004, 28, 1098–1102. [Google Scholar] [CrossRef]
- Cucchiari, D.; Bertuzzi, A.; Colombo, P.; De Sanctis, R.; Faucher, E.; Fusco, N.; Pellegrinelli, A.; Arosio, P.; Angelini, C. Juxtaglomerular cell tumor: Multicentric synchronous disease associated with paraneoplastic syndrome. J. Clin. Oncol. 2013, 31, e240–e242. [Google Scholar] [CrossRef]
- Shera, A.H.; Baba, A.A.; Bakshi, I.H.; Lone, I.A. Recurrent malignant juxtaglomerular cell tumor: A rare cause of malignant hypertension in a child. J. Indian Assoc. Pediatr. Surg. 2011, 16, 152–154. [Google Scholar] [PubMed]
- Dong, D.; Li, H.; Yan, W.; Xu, W. Juxtaglomerular cell tumor of the kidney--a new classification scheme. Urol. Oncol. 2010, 28, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.; Hsu, T.H.; Perlroth, M.G.; Hofmann, L.V.; Haynes, C.M.; Katznelson, L. Reninoma: Case report and literature review. J. Hypertens. 2008, 26, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Gottardo, F.; Cesari, M.; Morra, A.; Gardiman, M.; Fassina, A.; Dal Bianco, M. A kidney tumor in an adolescent with severe hypertension and hypokalemia: An uncommon case--case report and review of the literature on reninoma. Urol. Int. 2010, 85, 121–124. [Google Scholar] [CrossRef]
- Castelli, P.K.; Dillman, J.R.; Smith, E.A.; Vellody, R.; Cho, K.; Stanley, J.C. Imaging of renin-mediated hypertension in children. AJR Am. J. Roentgenol. 2013, 200, W661–W672. [Google Scholar] [CrossRef]
- Wolley, M.; Gordon, R.D.; Stowasser, M. Reninoma: The importance of renal vein renin ratios for lateralisation and diagnosis. Am. J. Nephrol. 2014, 39, 16–19. [Google Scholar] [CrossRef]
- Haab, F.; Duclos, J.M.; Guyenne, T.; Plouin, P.F.; Corvol, P. Renin secreting tumors: Diagnosis, conservative surgical approach and long-term results. J. Urol. 1995, 153, 1781–1784. [Google Scholar] [CrossRef]
- Niikura, S.; Komatsu, K.; Uchibayashi, T.; Ise, T.; Yokoyama, H.; Kobayashi, K.; Matsui, O.; Nonomura, A.; Namiki, M. Juxtaglomerular cell tumor of the kidney treated with nephron-sparing surgery. Urol. Int. 2000, 65, 160–162. [Google Scholar] [CrossRef]
- Martin, S.A.; Mynderse, L.A.; Lager, D.J.; Cheville, J.C. Juxtaglomerular cell tumor: A clinicopathologic study of four cases and review of the literature. Am. J. Clin. Pathol. 2001, 116, 854–863. [Google Scholar] [CrossRef] [Green Version]
- Mete, U.K.; Niranjan, J.; Kusum, J.; Rajesh, L.S.; Goswami, A.K.; Sharma, S.K. Reninoma treated with nephron-sparing surgery. Urology 2003, 61, 1259. [Google Scholar] [CrossRef]
- Xu, B.; Zhang, Q.; Jin, J. Hypertension secondary to reninoma treated with laparoscopic nephron-sparing surgery in a child. Urology 2012, 80, 210–213. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, N.; Gotoda, H.; Ohe, C.; Mikami, S.; Inoue, K.; Nagashima, Y.; Petersson, F.; Alvarado-Cabrero, I.; Pan, C.C.; Hes, O.; et al. Review of juxtaglomerular cell tumor with focus on pathobiological aspect. Diagn. Pathol. 2011, 6, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Kim, C.H.; Choi, Y.J.; Ayala, A.G.; Amirikachi, M.; Ro, J.Y. Juxtaglomerular cell tumor of kidney with CD34 and CD117 immunoreactivity: Report of 5 cases. Arch. Pathol. Lab. Med. 2006, 130, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, N.; Maris, S.; Monzon, F.A.; Tan, P.H.; Thomas, A.; Petersson, F.B.; Gatalica, Z.; Ghazalpour, A.; Bender, R.P.; Grossmann, P.; et al. Juxtaglomerular cell tumor: A morphological, immunohistochemical and genetic study of six cases. Hum. Pathol. 2013, 44, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Brandal, P.; Busund, L.T.; Heim, S. Chromosome abnormalities in juxtaglomerular cell tumors. Cancer 2005, 104, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Capovilla, M.; Couturier, J.; Molinié, V.; Amsellem-Ouazana, D.; Priollet, P.; Baumert, H.; Bruneval, P.; Vieillefond, A. Loss of chromosomes 9 and 11 may be recurrent chromosome imbalances in juxtaglomerular cell tumors. Hum. Pathol. 2008, 39, 459–462. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papez, J.; Starha, J.; Zerhau, P.; Pavlovska, D.; Jezova, M.; Jurencak, T.; Slaba, K.; Sterba, M.; Kerekes, A.; Merta, T.; et al. Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report. Genes 2021, 12, 220. https://doi.org/10.3390/genes12020220
Papez J, Starha J, Zerhau P, Pavlovska D, Jezova M, Jurencak T, Slaba K, Sterba M, Kerekes A, Merta T, et al. Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report. Genes. 2021; 12(2):220. https://doi.org/10.3390/genes12020220
Chicago/Turabian StylePapez, Jan, Jiri Starha, Pavel Zerhau, Denisa Pavlovska, Marta Jezova, Tomas Jurencak, Katerina Slaba, Martin Sterba, Arpad Kerekes, Tomas Merta, and et al. 2021. "Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report" Genes 12, no. 2: 220. https://doi.org/10.3390/genes12020220
APA StylePapez, J., Starha, J., Zerhau, P., Pavlovska, D., Jezova, M., Jurencak, T., Slaba, K., Sterba, M., Kerekes, A., Merta, T., Haluskova, T., Palova, H., Slaby, O., Sterba, J., & Jabandziev, P. (2021). Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report. Genes, 12(2), 220. https://doi.org/10.3390/genes12020220