Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report



{kind=link}
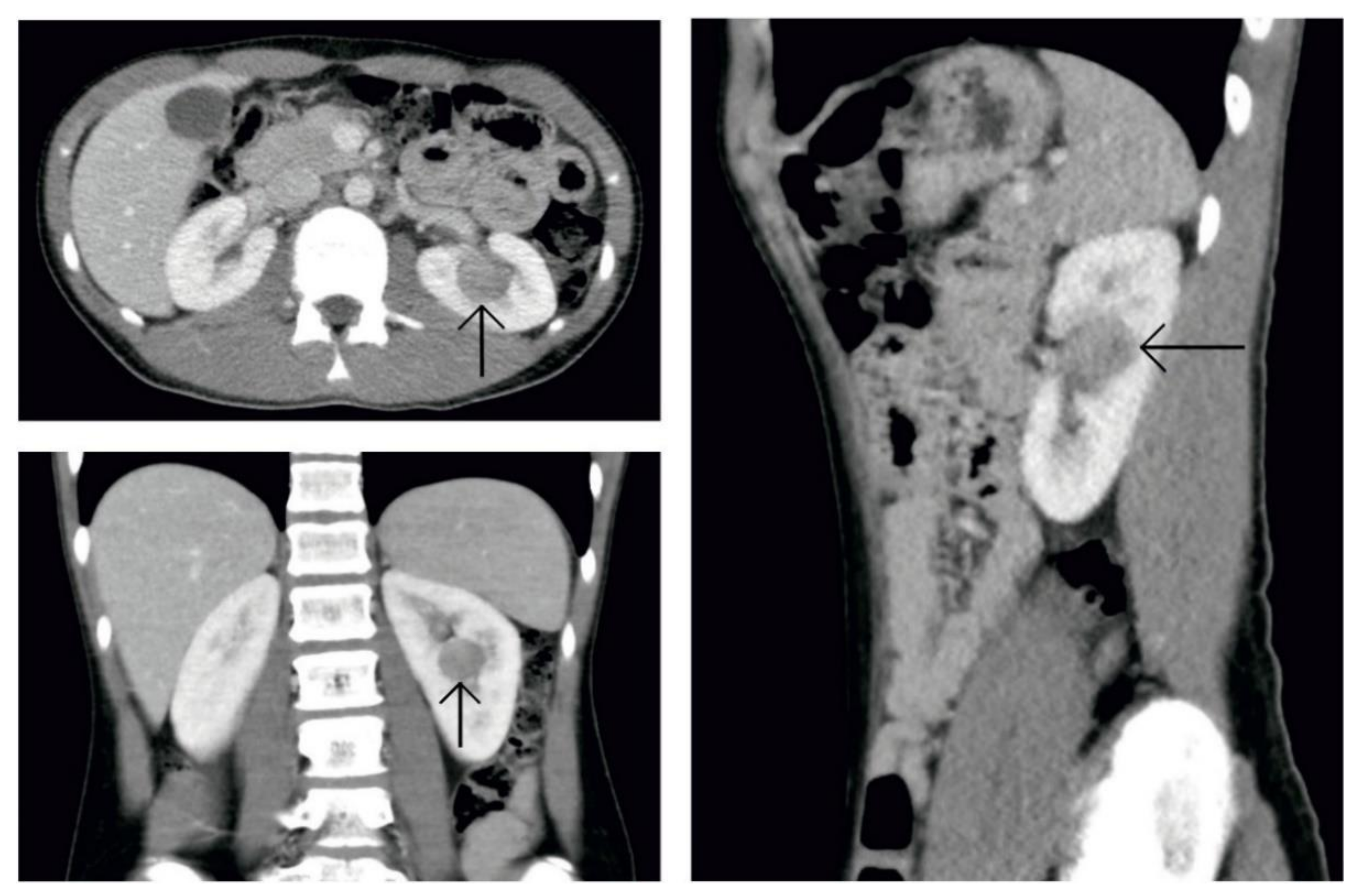{kind=link}
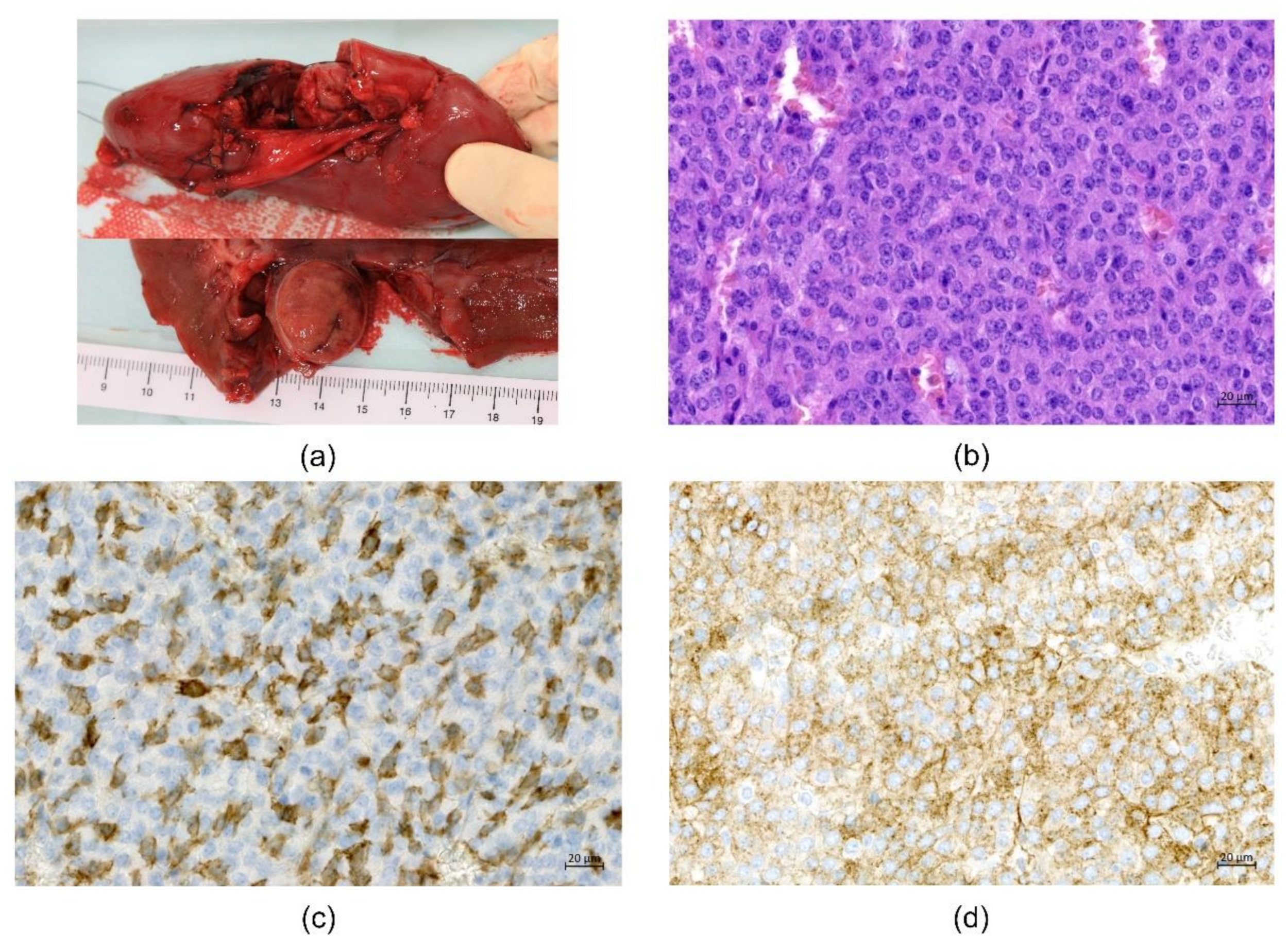{kind=link}
Abstract
Share and Cite
Papez, J.; Starha, J.; Zerhau, P.; Pavlovska, D.; Jezova, M.; Jurencak, T.; Slaba, K.; Sterba, M.; Kerekes, A.; Merta, T.; et al. Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report. Genes 2021, 12, 220. https://doi.org/10.3390/genes12020220
Papez J, Starha J, Zerhau P, Pavlovska D, Jezova M, Jurencak T, Slaba K, Sterba M, Kerekes A, Merta T, et al. Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report. Genes. 2021; 12(2):220. https://doi.org/10.3390/genes12020220
Chicago/Turabian StylePapez, Jan, Jiri Starha, Pavel Zerhau, Denisa Pavlovska, Marta Jezova, Tomas Jurencak, Katerina Slaba, Martin Sterba, Arpad Kerekes, Tomas Merta, and et al. 2021. "Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report" Genes 12, no. 2: 220. https://doi.org/10.3390/genes12020220
APA StylePapez, J., Starha, J., Zerhau, P., Pavlovska, D., Jezova, M., Jurencak, T., Slaba, K., Sterba, M., Kerekes, A., Merta, T., Haluskova, T., Palova, H., Slaby, O., Sterba, J., & Jabandziev, P. (2021). Spindle Cell Hemangioma and Atypically Localized Juxtaglomerular Cell Tumor in a Patient with Hereditary BRIP1 Mutation: A Case Report. Genes, 12(2), 220. https://doi.org/10.3390/genes12020220