
Estimating Copy-Number Proportions: The Comeback of Sanger Sequencing


Abstract
:1. Introduction
2. Single-Nucleotide Polymorphisms (SNPs) and Indels
3. Base Editing by CRISPR-Cas9 Endonucleases and Nickases
4. C Methylation
5. RNA Editing
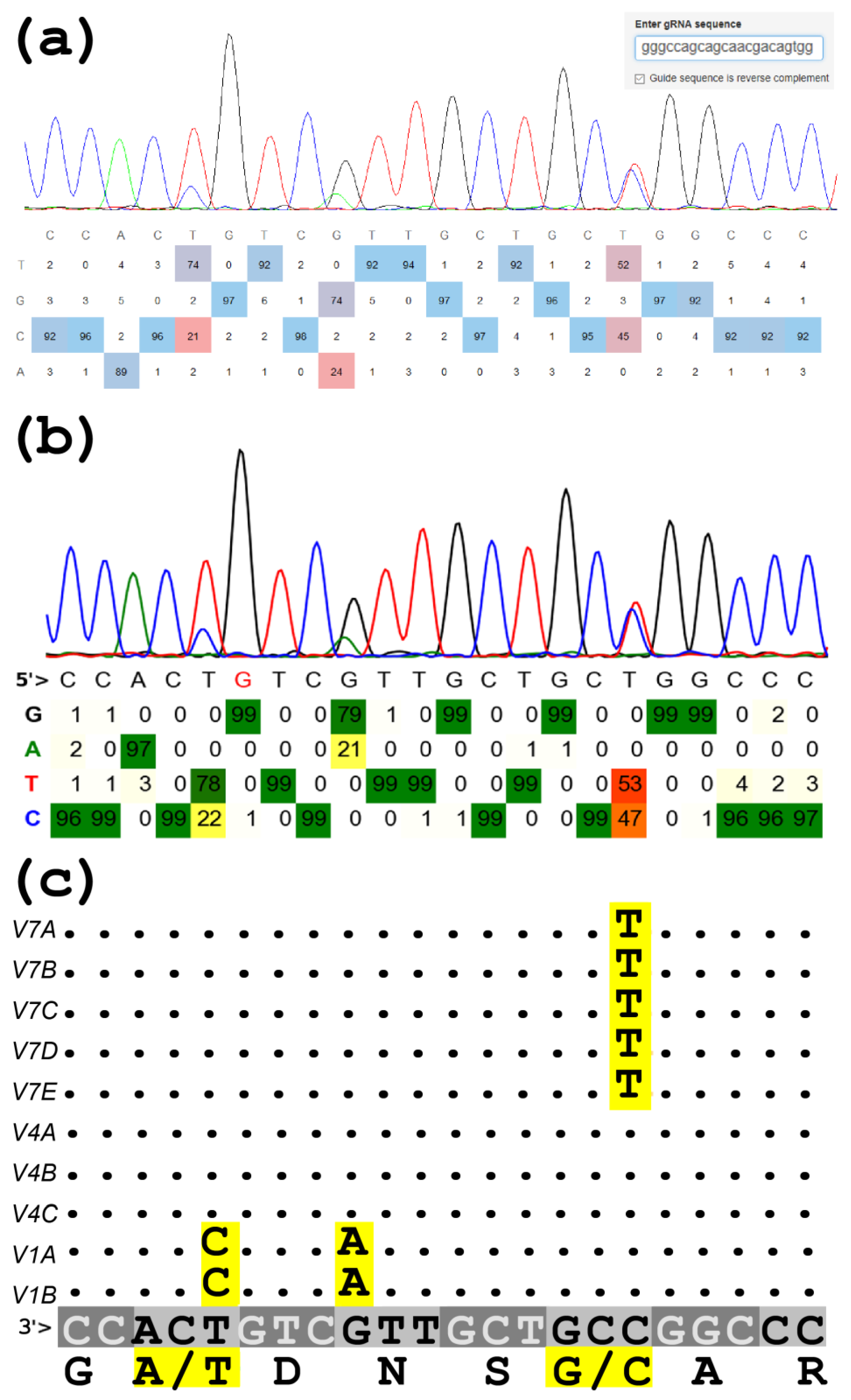
6. Copy-Number Variations (CNVs)
7. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanger, F.; Coulson, A.R. A rapid method for determining sequences in DNA by primed synthesis with DNA polymerase. J. Mol. Biol. 1975, 94, 441–448. [Google Scholar] [CrossRef]
- Ishino, S.; Ishino, Y. DNA polymerases as useful reagents for biotechnology—The history of developmental research in the field. Front. Microbiol. 2014, 5, 465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, L.T.; Zakeri, H.; Deng, Q.; Spurgeon, S.; Kwok, P.Y.; Nickerson, D.A. AmpliTaq(R) DNA polymerase, FS dye-terminator sequencing: Analysis of peak height patterns. Biotechniques 1996, 21, 694–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Horn, P.B.; Davis, M.C.; Cunniff, J.J.; Ruan, C.; McArdle, B.F.; Samols, S.B.; Szasz, J.; Hu, G.; Hujer, K.M.; Domke, S.T.; et al. Thermo Sequenase DNA polymerase and T. acidophilum pyrophosphatase: New thermostable enzymes for DNA sequencing. Biotechniques 1997, 22, 758–762. [Google Scholar] [CrossRef]
- Tabor, S.; Richardson, C.C. DNA sequence analysis with a modified bacteriophage T7 DNA polymerase. Effect of pyrophosphorolysis and metal ions. J. Biol. Chem. 1990, 265, 8322–8328. [Google Scholar] [CrossRef]
- Korch, C.; Drabkin, H. Improved DNA sequencing accuracy and detection of heterozygous alleles using manganese citrate and different fluorescent dye terminators. Genome Res. 1999, 9, 588–595. [Google Scholar]
- Jiang, M.; Zhang, Y.; Fei, J.; Chang, X.; Fan, W.; Qian, X.; Zhang, T.; Lu, D. Rapid quantification of DNA methylation by measuring relative peak heights in direct bisulfite-PCR sequencing traces. Lab. Investig. 2009, 90, 282–290. [Google Scholar] [CrossRef]
- Shen, W.; Tian, Y.; Ran, T.; Gao, Z.Q. Genotyping and quantification techniques for single-nucleotide polymorphisms. Trac-Trends Anal. Chem. 2015, 69, 1–13. [Google Scholar] [CrossRef]
- Lefever, S.; Rihani, A.; Van der Meulen, J.; Pattyn, F.; Van Maerken, T.; Van Dorpe, J.; Hellemans, J.; Vandesompele, J. Cost-effective and robust genotyping using double-mismatch allele-specific quantitative PCR. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Roca, I.; Gonzalez-Castro, L.; Fernandez, H.; Couce, M.L.; Fernandez-Marmiesse, A. Free-access copy-number variant detection tools for targeted next-generation sequencing data. Mutat. Res. Rev. Mutat. Res. 2019, 779, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Taillon-Miller, P.; Piernot, E.E.; Kwok, P.Y. Efficient approach to unique single-nucleotide polymorphism discovery. Genome Res. 1999, 9, 499–505. [Google Scholar] [PubMed]
- Humma, L.M.; Farmerie, W.G.; Wallace, M.R.; Johnson, J.A. Sequencing of beta 2-adrenoceptor gene PCR products using Taq BigDye terminator chemistry results in inaccurate base calling. Biotechniques 2000, 29, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirak, A.; Seroussi, U.; Gootwine, E.; Seroussi, E. Sequence motifs capable of forming DNA stem-loop structures act as a replication diode. FEBS Open Bio. 2017, 7, 944–952. [Google Scholar] [CrossRef]
- Carr, I.M.; Robinson, J.I.; Dimitriou, R.; Markham, A.F.; Morgan, A.W.; Bonthron, D.T. Inferring relative proportions of DNA variants from sequencing electropherograms. Bioinformatics 2009, 25, 3244–3250. [Google Scholar] [CrossRef] [PubMed]
- McCluskey, M.; Ni, S.; Hulce, D.; Liu, J. DNA Mutation and Methylation Quantification from Sanger Sequencing Traces with Mutation Surveyor Software. Available online: https://softgenetics.com/PDF/MutationSurveyorQuantification.pdf (accessed on 16 February 2021).
- Seroussi, E.; Ron, M.; Kedra, D. ShiftDetector: Detection of shift mutations. Bioinformatics 2002, 18, 1137–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.T.; Demarest, B.L.; Bisgrove, B.W.; Su, Y.C.; Smith, M.; Yost, H.J. Poly peak parser: Method and software for identification of unknown indels using sanger sequencing of polymerase chain reaction products. Dev. Dyn. 2014, 243, 1632–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhidkov, I.; Cohen, R.; Geifman, N.; Mishmar, D.; Rubin, E. CHILD: A new tool for detecting low-abundance insertions and deletions in standard sequence traces. Nucleic Acids Res. 2010, 39, e47. [Google Scholar] [CrossRef] [Green Version]
- Sobenin, I.A.; Mitrofanov, K.Y.; Zhelankin, A.V.; Sazonova, M.A.; Postnov, A.Y.; Revin, V.V.; Bobryshev, Y.V.; Orekhov, A.N. Quantitative assessment of heteroplasmy of mitochondrial genome: Perspectives in diagnostics and methodological pitfalls. Biomed. Res. Int. 2014, 2014, 292017. [Google Scholar] [CrossRef]
- Duan, M.; Tu, J.; Lu, Z. Recent Advances in Detecting Mitochondrial DNA Heteroplasmic Variations. Molecules 2018, 23, 323. [Google Scholar] [CrossRef] [Green Version]
- Blazej, R.G.; Paegel, B.M.; Mathies, R.A. Polymorphism ratio sequencing: A new approach for single nucleotide polymorphism discovery and genotyping. Genome. Res. 2003, 13, 287–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rohlin, A.; Wernersson, J.; Engwall, Y.; Wiklund, L.; Bjoerk, J.; Nordling, M. Parallel sequencing used in detection of mosaic mutations: Comparison with four diagnostic DNA screening techniques. Hum. Mutat. 2009, 30, 1012–1020. [Google Scholar] [CrossRef] [PubMed]
- Naue, J.; Sanger, T.; Schmidt, U.; Klein, R.; Lutz-Bonengel, S. Factors affecting the detection and quantification of mitochondrial point heteroplasmy using Sanger sequencing and SNaPshot minisequencing. Int. J. Legal Med. 2011, 125, 427–436. [Google Scholar] [CrossRef]
- Zhang, S.; Shen, J.; Li, D.; Cheng, Y. Strategies in the delivery of Cas9 ribonucleoprotein for CRISPR/Cas9 genome editing. Theranostics 2021, 11, 614–648. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Prot. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]
- Kluesner, M.G.; Nedveck, D.A.; Lahr, W.S.; Garbe, J.R.; Abrahante, J.E.; Webber, B.R.; Moriarity, B.S. EditR: A Method to quantify base editing from Sanger sequencing. CRISPR J. 2018, 1, 239–250. [Google Scholar] [CrossRef] [PubMed]
- Hsiau, T.; Maures, T.; Waite, K.; Yang, J.; Kelso, R.; Holden, K.; Stoner, R. Inference of CRISPR edits from Sanger trace data. bioRxiv 2019, 251082. [Google Scholar]
- Chatterjee, P.; Jakimo, N.; Jacobson, J.M. Minimal PAM specificity of a highly similar SpCas9 ortholog. Sci. Adv. 2018, 4, eaau0766. [Google Scholar] [CrossRef] [Green Version]
- Xu, L.; Liu, Y.; Han, R. BEAT: A Python program to quantify base editing from Sanger sequencing. CRISPR J. 2019, 2, 223–229. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kluesner, M.G.; Arnold, A.; Lerner, T.; Tasakis, R.N.; Wüst, S.; Binder, M.; Moriarity, B.S. MultiEditR: An easy validation method for detecting and quantifying RNA editing from Sanger sequencing. bioRxiv 2019, 633685. [Google Scholar]
- Wreczycka, K.; Gosdschan, A.; Yusuf, D.; Gruning, B.; Assenov, Y.; Akalin, A. Strategies for analyzing bisulfite sequencing data. J. Biotechnol. 2017, 261, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Pajares, M.J.; Palanca-Ballester, C.; Urtasun, R.; Alemany-Cosme, E.; Lahoz, A.; Sandoval, J. Methods for analysis of specific DNA methylation status. Methods 2020, in press. [Google Scholar]
- Toung, J.M.; Lahens, N.; Hogenesch, J.B.; Grant, G. Detection theory in identification of RNA-DNA sequence differences using RNA-sequencing. PLoS ONE 2014, 9, e112040. [Google Scholar] [CrossRef] [Green Version]
- Oakes, E.; Vadlamani, P.; Hundley, H.A. Methods for the detection of adenosine-to-inosine editing events in cellular RNA. In mRNA Processing: Methods and Protocols; Shi, Y., Ed.; Springer: New York, NY, USA, 2017; pp. 103–127. [Google Scholar]
- Feuk, L.; Carson, A.R.; Scherer, S.W. Structural variation in the human genome. Nat. Rev. Genet. 2006, 7, 85–97. [Google Scholar] [CrossRef]
- Seroussi, E.; Klompus, S.; Silanikove, M.; Krifucks, O.; Shapiro, F.; Gertler, A.; Leitner, G. Nonbactericidal secreted phospholipase A2s are potential anti-inflammatory factors in the mammary gland. Immunogenetics 2013, 65, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Curzon, A.Y.; Shirak, A.; Dor, L.; Zak, T.; Perelberg, A.; Seroussi, E.; Ron, M. A duplication of the Anti-Mullerian hormone gene is associated with genetic sex determination of different Oreochromis niloticus strains. Heredity (Edinb) 2020, 125, 317–327. [Google Scholar] [CrossRef]
- Seroussi, E.; Blum, S.E.; Krifucks, O.; Shirak, A.; Jacoby, S.; Leitner, G. Basal levels of CD18 antigen presenting cells in cow milk associate with copy-number variation of Fc Gamma Receptors. Genes (Basel) 2020, 11, 952. [Google Scholar] [CrossRef]
- Staden, R.; Beal, K.F.; Bonfield, J.K. The Staden package, 1998. Methods Mol. Biol. 2000, 132, 115–130. [Google Scholar]
- Tillett, D. In Depth: Mixed Basecalling and Simple Mixed Basecalling. Available online: http://cowry.agri.huji.ac.il/InDepthMixedBasecalling.htm (accessed on 16 February 2021).
- Zouros, E. Biparental Inheritance through uniparental transmission: The doubly uniparental inheritance (DUI) of mitochondrial DNA. Evol. Biol. 2013, 40, 1–31. [Google Scholar] [CrossRef]
- Allex, C.F.; Shavlik, J.W.; Blattner, F.R. Neural network input representations that produce accurate consensus sequences from DNA fragment assemblies. Bioinformatics 1999, 15, 723–728. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Software | Implemented in | Use Focus | Download/Tool Site | Web Tool | Reference |
|---|---|---|---|---|---|
| QSVanalyzer | VB.Net | Single-Nucleotide Polymorphisms (SNPs) | http://dna-leeds.co.uk/qsv/download.php (accessed on 16 February 2021) | N | [14] |
| BioEdit | C++ | SNPs | https://bioedit.software.informer.com/ (accessed on 16 February 2021) | N | [31] |
| Chromas | C++ | SNPs | http://technelysium.com.au/wp/chromas/ (accessed on 16 February 2021) | N | Freemium |
| TIDE/TIDER | R | CRISPR indels | https://tide.nki.nl/ (accessed on 16 February 2021) | Y | [26] |
| ICE | Python | CRISPR indels | https://ice.synthego.com/#/ (accessed on 16 February 2021) | Y | [28] |
| EditR | R | CRISPR SNPs | https://moriaritylab.shinyapps.io/editr_v10/ (accessed on 16 February 2021) | Y | [27] |
| BEEP | Python | CRISPR indels | https://github.com/mitmedialab/BEEP (accessed on 16 February 2021) | N | [29] |
| BEAT | Python | CRISPR SNPs | https://hanlab.cc/beat/ (accessed on 16 February 2021) | Y | [30] |
| MultiEditR | R | RNA SNPs | https://moriaritylab.shinyapps.io/multieditr/ (accessed on 16 February 2021) | Y | [32] |
| Base String 1 | G Peak Height | Base String 1 | C Peak Height |
|---|---|---|---|
| GCG | small | GCC | average/large |
| TCG | small | TCC | ND |
| CCG | ND | CCC | ND |
| ACG | small | ACC | average/large |
| Variants | Expected | EditR | BEAT | Chromas/BioEdit | QSV |
|---|---|---|---|---|---|
| T/C | 4 | 3.52 | 3.55 | 3.50 | 3.54 |
| G/A | 4 | 3.08 | 3.79 | 3.08 | 3.03 |
| C/T | 1 | 0.87 | 0.89 | 0.87 | 0.87 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seroussi, E. Estimating Copy-Number Proportions: The Comeback of Sanger Sequencing. Genes 2021, 12, 283. https://doi.org/10.3390/genes12020283
Seroussi E. Estimating Copy-Number Proportions: The Comeback of Sanger Sequencing. Genes. 2021; 12(2):283. https://doi.org/10.3390/genes12020283
Chicago/Turabian StyleSeroussi, Eyal. 2021. "Estimating Copy-Number Proportions: The Comeback of Sanger Sequencing" Genes 12, no. 2: 283. https://doi.org/10.3390/genes12020283
APA StyleSeroussi, E. (2021). Estimating Copy-Number Proportions: The Comeback of Sanger Sequencing. Genes, 12(2), 283. https://doi.org/10.3390/genes12020283