
Pancytopenia, Recurrent Infection, Poor Wound Healing, Heterotopia of the Brain Probably Associated with A Candidate Novel de Novo CDC42 Gene Defect: Expanding the Molecular and Phenotypic Spectrum

 ,
,  , ,
, , 
 , ,
, ,  , , , add
Show full author list
, , , add
Show full author list
Abstract
:1. Background
2. Materials and Methods
2.1. Human Subjects
2.2. Ethical Approval
2.3. DNA Extraction
2.4. Whole Exome Sequencing (WES)
2.5. Bioinformatics Analysis
2.6. Mutation Confirmation and Sequencing Analysis
2.7. Cell Isolation and Culture
2.8. Cell Treatment
2.9. Wound Healing Assay
2.10. Cell Viability Assay
2.11. Cell Cycle Assay
2.12. Protein Modeling
2.13. Statistical Analysis
3. Results
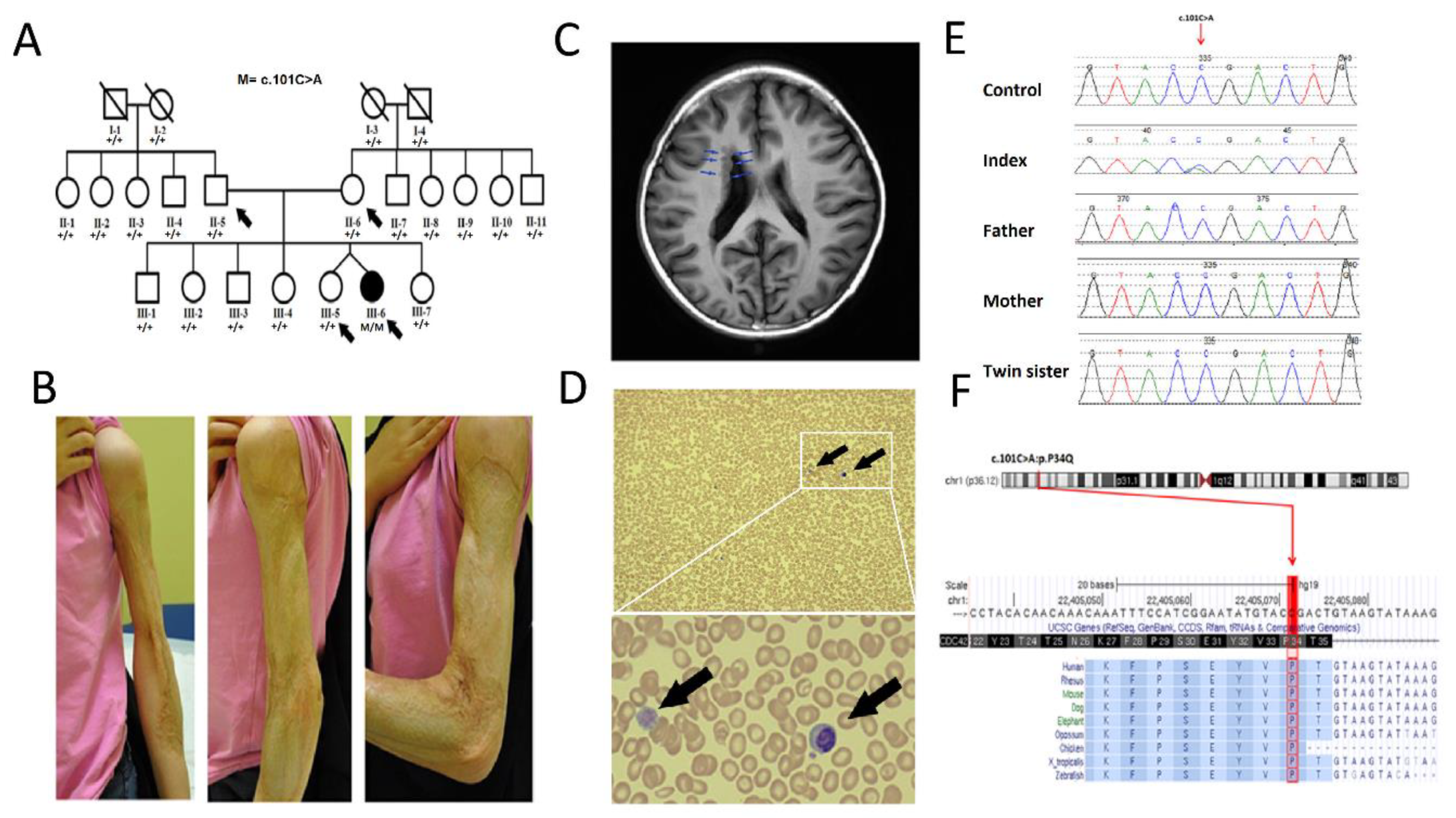
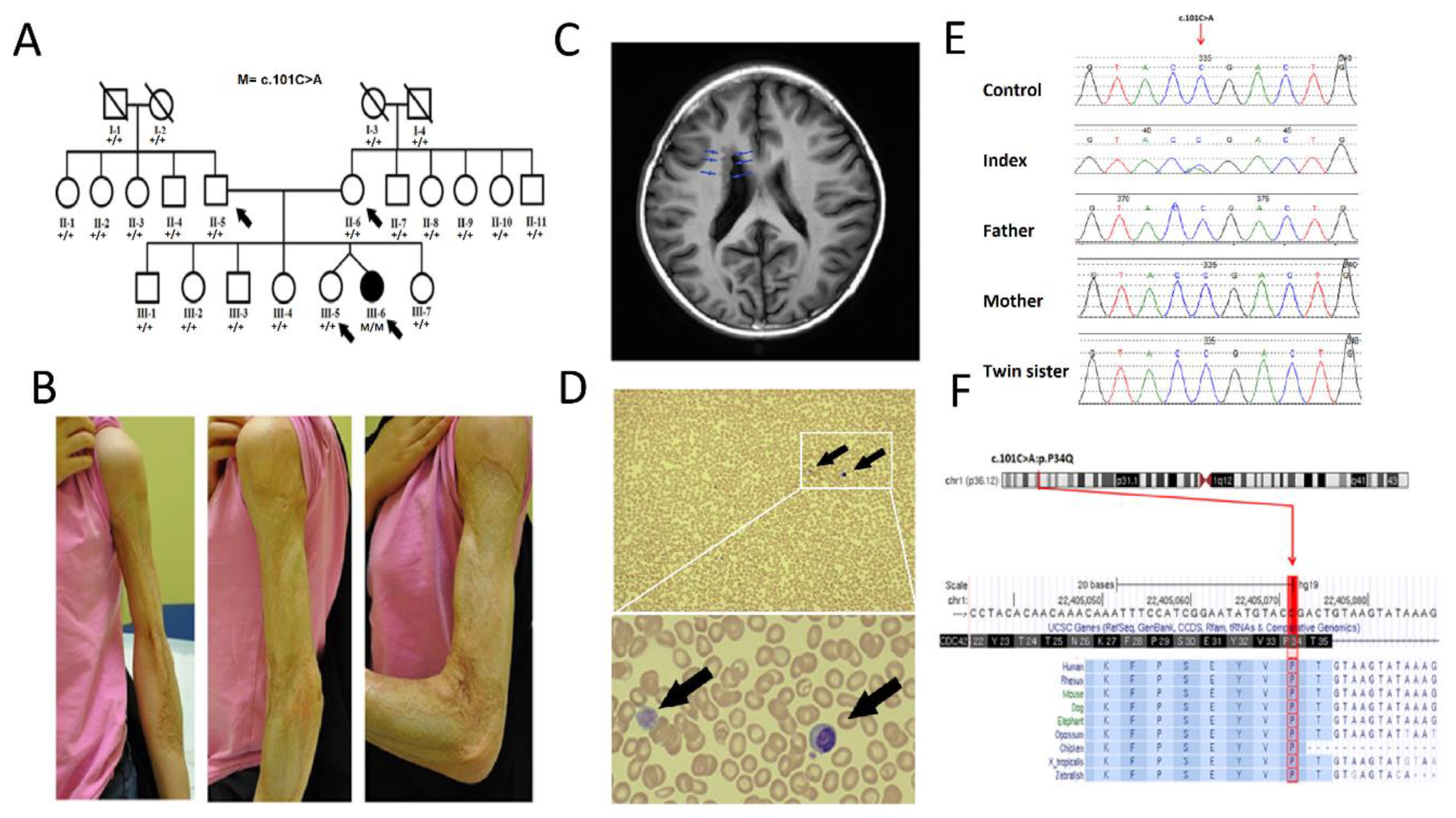
3.1. Clinical Description
3.2. Genetic Analysis
3.3. Functional Studies
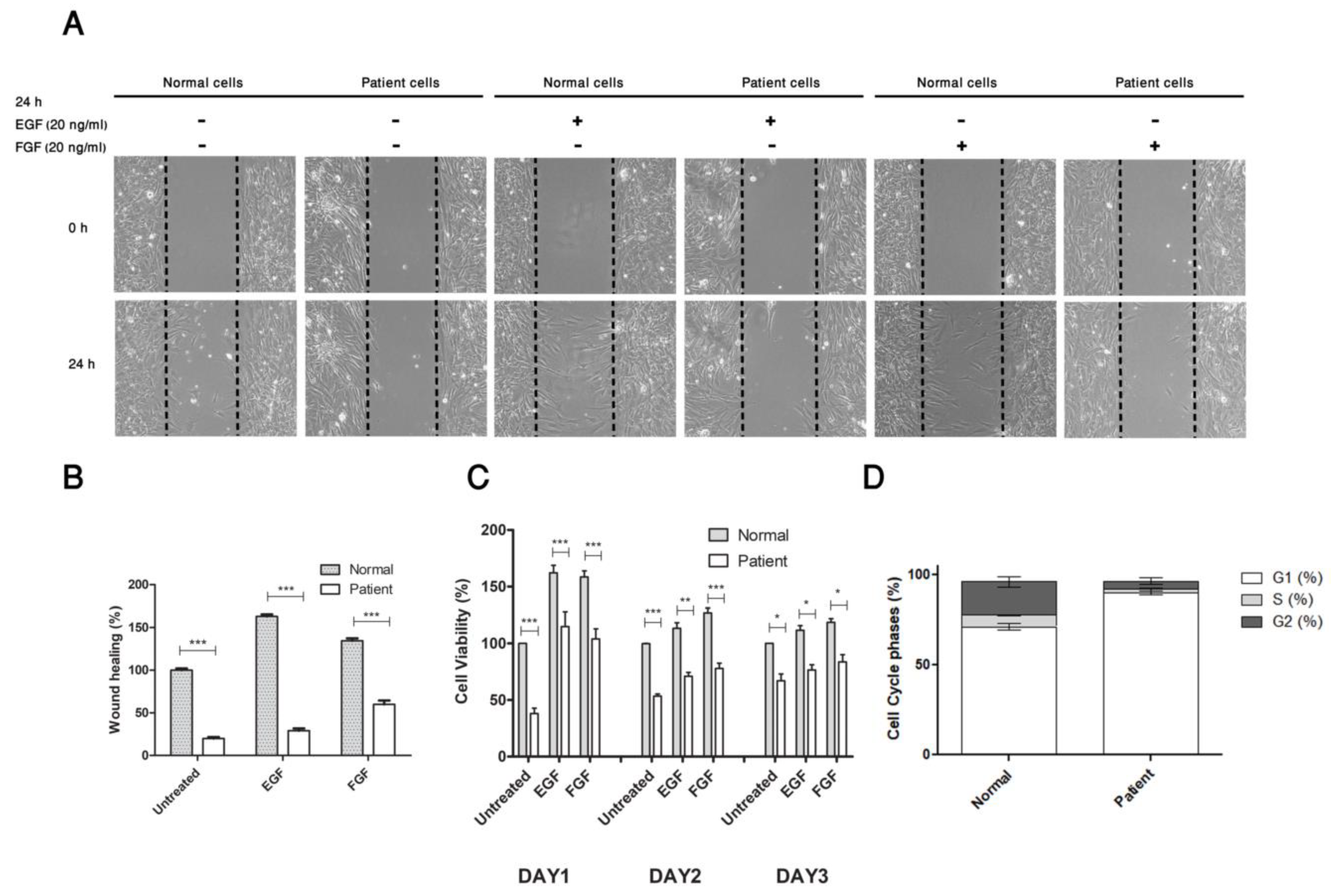
3.3.1. Mutated CDC42 Selectively Abrogates EGF/FGF Induced Cell Migration and Proliferation
3.3.2. Substitution of Pro to Gln at Position 34 Induces Arrest in G1 Phase of Patient’s Cell Cycle Progression
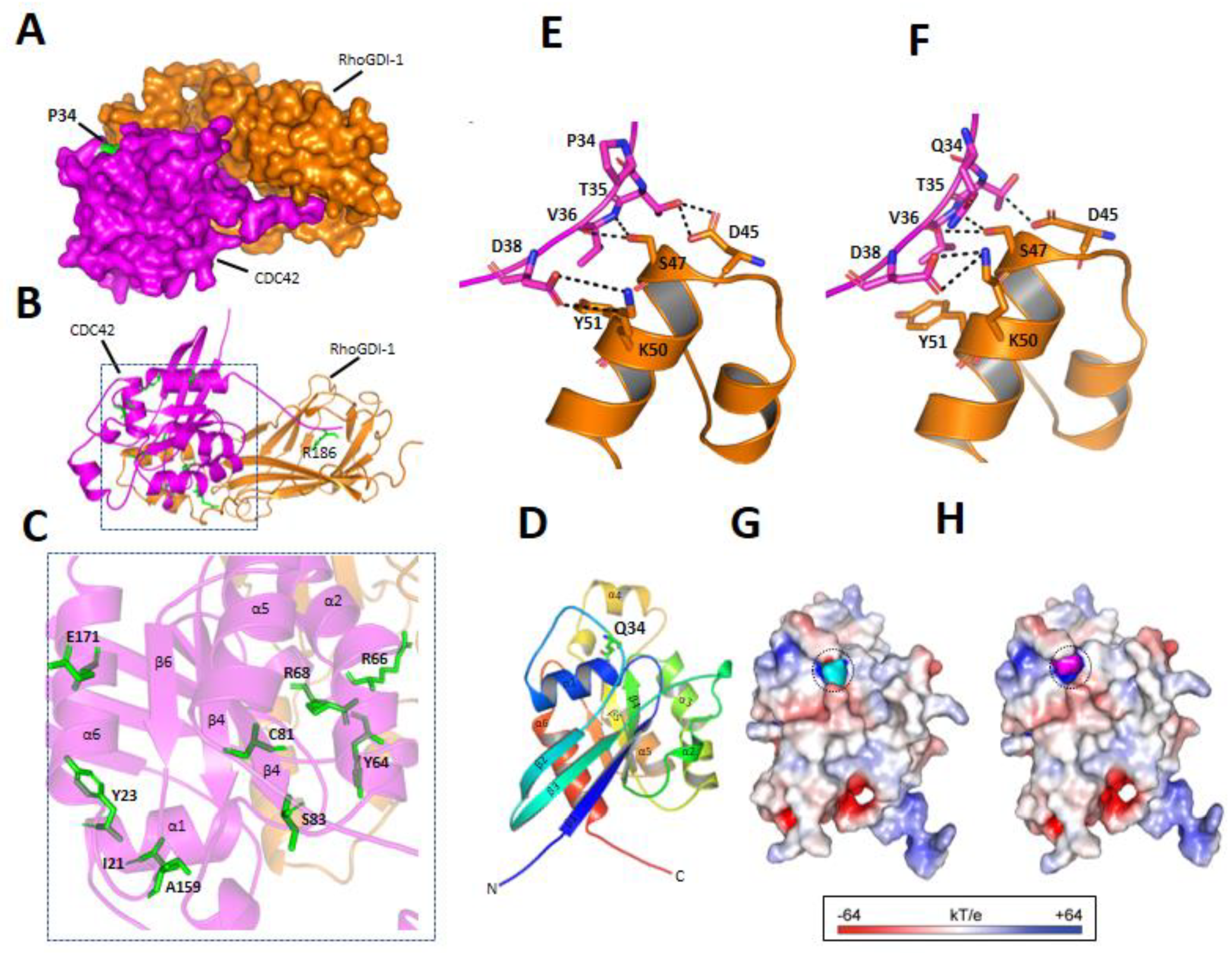
3.3.3. p.P34Q Mutation Affects the Binding of CDC42 to RhoGDI-1 Protein
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yamaguchi, Y.; Yoshikawa, K. Cutaneous Wound Healing: An Update. J. Dermatol. 2001, 28, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Reinke, J.; Sorg, H. Wound Repair and Regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef]
- Rumalla, V.K.; Borah, G.L. Cytokines, Growth Factors, and Plastic Surgery. Plast. Reconstr. Surg. 2001, 108, 719–733. [Google Scholar] [CrossRef]
- Barrientos, S.; Stojadinovic, O.; Golinko, M.S.; Brem, H.; Tomic-Canic, M. PERSPECTIVE ARTICLE: Growth factors and cytokines in wound healing. Wound Repair Regen. 2008, 16, 585–601. [Google Scholar] [CrossRef]
- Etienne-Manneville, S.; Hall, A. Rho GTPases in cell biology. Nat. Cell Biol. 2002, 420, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Pothula, S.; Bazan, H.E.P.; Chandrasekher, G. Regulation of Cdc42 Expression and Signaling Is Critical for Promoting Corneal Epithelial Wound Healing. Investig. Opthalmol. Vis. Sci. 2013, 54, 5343–5352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Yang, W. Cellular signaling for activation of Rho GTPase Cdc42. Cell. Signal. 2008, 20, 1927–1934. [Google Scholar] [CrossRef]
- Takenouchi, T.; Kosaki, R.; Niizuma, T.; Hata, K.; Kosaki, K. Macrothrombocytopenia and developmental delay with a de novo CDC42 mutation: Yet another locus for thrombocytopenia and developmental delay. Am. J. Med Genet. Part A 2015, 167, 2822–2825. [Google Scholar] [CrossRef] [PubMed]
- Takenouchi, T.; Okamoto, N.; Ida, S.; Uehara, T.; Kosaki, K. Further evidence of a mutation in CDC42 as a cause of a recognizable syndromic form of thrombocytopenia. Am. J. Med Genet. Part A 2015, 170, 852–855. [Google Scholar] [CrossRef]
- Motokawa, M.; Watanabe, S.; Nakatomi, A.; Kondoh, T.; Matsumoto, T.; Morifuji, K.; Sawada, H.; Nishimura, T.; Nunoi, H.; Yoshiura, K.-I.; et al. A hot-spot mutation in CDC42 (p.Tyr64Cys) and novel phenotypes in the third patient with Takenouchi-Kosaki syndrome. J. Hum. Genet. 2018, 63, 387–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinelli, S.; Krumbach, O.H.; Pantaleoni, F.; Coppola, S.; Amin, E.; Pannone, L.; Nouri, K.; Farina, L.; Dvorsky, R.; Lepri, F.; et al. Functional Dysregulation of CDC42 Causes Diverse Developmental Phenotypes. Am. J. Hum. Genet. 2018, 102, 309–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Lv, J.; Wang, P.; Liao, Y.; Li, Y.; Zhao, W.; Zen, J.; Dong, Z.; Guo, Z.; Bo, X.; et al. Vascular endothelial Cdc42 deficiency delays skin wound-healing processes by increasing IL-1β and TNF-α expression. Am. J. Transl. Res. 2019, 11, 257–268. [Google Scholar]
- Alfares, A.; Alfadhel, M.; Mujamammi, A.; Alotaibi, B.; Albahkali, S.; Al Balwi, M.; Benabdelkamel, H.; Masood, A.; Ali, R.; Almuaysib, A.; et al. Proteomic and Molecular Assessment of the Common Saudi Variant in ACADVL Gene Through Mesenchymal Stem Cells. Front. Cell Dev. Biol. 2020, 7, 365. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, G.R.; Nassar, N.; Cerione, R.A. Structure of the Rho Family GTP-Binding Protein Cdc42 in Complex with the Multifunctional Regulator RhoGDI. Cell 2000, 100, 345–356. [Google Scholar] [CrossRef] [Green Version]
- Lam, M.T.; Coppola, S.; Krumbach, O.H.; Prencipe, G.; Insalaco, A.; Cifaldi, C.; Brigida, I.; Zara, E.; Scala, S.; Di Cesare, S.; et al. A novel disorder involving dyshematopoiesis, inflammation, and HLH due to aberrant CDC42 function. J. Exp. Med. 2019, 216, 2778–2799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczawinska-Poplonyk, A.; Ploski, R.; Bernatowska, E.; Pac, M. A Novel CDC42 Mutation in an 11-Year Old Child Manifesting as Syndromic Immunodeficiency, Autoinflammation, Hemophagocytic Lymphohistiocytosis, and Malignancy: A Case Report. Front. Immunol. 2020, 11, 318. [Google Scholar] [CrossRef] [Green Version]
- Colicelli, J. Human RAS Superfamily Proteins and Related GTPases. Sci. Signal. 2004, 2004, re13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvorský, R.; Ahmadian, M.R. Always look on the bright site of Rho: Structural implications for a conserved intermolecular interface. EMBO Rep. 2004, 5, 1130–1136. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, L.; Geiger, H.; Cancelas, J.A.; Mo, J.; Zheng, Y. Rho GTPase Cdc42 coordinates hematopoietic stem cell quiescence and niche interaction in the bone marrow. Proc. Natl. Acad. Sci. USA 2007, 104, 5091–5096. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, L.; Kalfa, T.A.; Cancelas, J.A.; Shang, X.; Pushkaran, S.; Mo, J.; Williams, D.A.; Zheng, Y. Cdc42 critically regulates the balance between myelopoiesis and erythropoiesis. Blood 2007, 110, 3853–3861. [Google Scholar] [CrossRef] [Green Version]
- Bucciol, G.; Pillay, B.; Casas-Martin, J.; Delafontaine, S.; Proesmans, M.; Lorent, N.; Coolen, J.; Tousseyn, T.; Bossuyt, X.; Ma, C.S.; et al. Systemic Inflammation and Myelofibrosis in a Patient with Takenouchi-Kosaki Syndrome due to CDC42 Tyr64Cys Mutation. J. Clin. Immunol. 2020, 40, 567–570. [Google Scholar] [CrossRef]
- Li, T.-T.; Li, J.; Geng, Y.-H.; Zhang, F.; Liu, L.; Yang, Y.-L. NRAS Gene Expression and Its Clinical Significance in Patients with Acute Myeloid Leukemia. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2020, 28, 76–81. [Google Scholar]
- Ridley, A.J.; Schwartz, M.A.; Burridge, K.; Firtel, R.A.; Ginsberg, M.H.; Borisy, G.; Parsons, J.T.; Horwitz, A.R. Cell Migration: Integrating Signals from Front to Back. Science 2003, 302, 1704–1709. [Google Scholar] [CrossRef] [Green Version]
- Fukata, M.; Nakagawa, M.; Kaibuchi, K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr. Opin. Cell Biol. 2003, 15, 590–597. [Google Scholar] [CrossRef]
- Raftopoulou, M.; Hall, A. Cell migration: Rho GTPases lead the way. Dev. Biol. 2004, 265, 23–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieck, P.W.; Cholidis, S.; Hartmann, C. Intracellular Signaling Pathway of FGF-2-modulated Corneal Endothelial Cell Migration during Wound Healing in vitro. Exp. Eye Res. 2001, 73, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Joyce, N.C.; Meklir, B. Protein kinase C activation during corneal endothelial wound repair. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1958–1973. [Google Scholar]
- Lee, J.G.; Kay, E.P. FGF-2-induced wound healing in corneal endothelial cells requires Cdc42 activation and Rho inactivation through the phosphatidylinositol 3-kinase pathway. Investig. Ophthalmol. Vis. Sci. 2006, 47, 1376–1386. [Google Scholar] [CrossRef] [Green Version]
- El-Sibai, M.; Nalbant, P.; Pang, H.; Flinn, R.J.; Sarmiento, C.; Macaluso, F.; Cammer, M.; Condeelis, J.S.; Hahn, K.M.; Backer, J.M. Cdc42 is required for EGF-stimulated protrusion and motility in MTLn3 carcinoma cells. J. Cell Sci. 2007, 120, 3465–3474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verboon, J.M.; Mahmut, D.; Kim, A.R.; Nakamura, M.; Abdulhay, N.J.; Nandakumar, S.K.; Gupta, N.; Akie, T.E.; Geddis, A.E.; Manes, B.; et al. Infantile Myelofibrosis and Myeloproliferation with CDC42 Dysfunction. J. Clin. Immunol. 2020, 40, 554–566. [Google Scholar] [CrossRef] [Green Version]
- Gernez, Y.; De Jesus, A.A.; Alsaleem, H.; Macaubas, C.; Roy, A.; Lovell, D.; Jagadeesh, K.A.; Alehashemi, S.; Erdman, L.; Grimley, M.; et al. Severe autoinflammation in 4 patients with C-terminal variants in cell division control protein 42 homolog (CDC42) successfully treated with IL-1β inhibition. J. Allergy Clin. Immunol. 2019, 144, 1122–1125.e6. [Google Scholar] [CrossRef] [Green Version]
- Gibson, R.M.; Wilson-Delfosse, A.L. RhoGDI-binding-defective mutant of Cdc42Hs targets to membranes and activates filopodia formation but does not cycle with the cytosol of mammalian cells. Biochem. J. 2001, 359, 285–294. [Google Scholar] [CrossRef]
- Swart-Mataraza, J.M.; Li, Z.; Sacks, D.B. IQGAP1 Is a Component of Cdc42 Signaling to the Cytoskeleton. J. Biol. Chem. 2002, 277, 24753–24763. [Google Scholar] [CrossRef] [Green Version]
- Gibson, R.M.; Gandhi, P.N.; Tong, X.; Miyoshi, J.; Takai, Y.; Konieczkowski, M.; Sedor, J.R.; Wilson-Delfosse, A.L. An activating mutant of Cdc42 that fails to interact with Rho GDP-dissociation inhibitor localizes to the plasma membrane and mediates actin reorganization. Exp. Cell Res. 2004, 301, 211–222. [Google Scholar] [CrossRef]
- Dasouki, M.; Okonkwo, K.C.; Ray, A.; Folmsbeel, C.K.; Gozales, D.; Keles, S.; Puck, J.M.; Chatila, T. Deficient T Cell Receptor Excision Circles (TRECs) in autosomal recessive hyper IgE syndrome caused by DOCK8 mutation: Implications for pathogenesis and potential detection by newborn screening. Clin. Immunol. 2011, 141, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Sun, X.; Wang, J.; Zhao, Q.; Dai, R.; Wang, Y.; Zhou, L.; Westerberg, L.; Ding, Y.; Zhao, X.; et al. Defective thymic output in WAS patients is associated with abnormal actin organization. Sci. Rep. 2017, 7, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Brigida, I.; Zoccolillo, M.; Cicalese, M.P.; Pfajfer, L.; Barzaghi, F.; Scala, S.; Oleaga-Quintas, C.; Álvarez-Álvarez, J.A.; Sereni, L.; Giannelli, S.; et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood 2018, 132, 2362–2374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michaelson, D.; Silletti, J.; Murphy, G.; D’Eustachio, P.; Rush, M.; Philips, M.R. Differential localization of Rho GTPases in live cells: Regulation by hypervariable regions and RhoGDI binding. J. Cell Biol. 2001, 152, 111–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueyama, T. Rho-Family Small GTPases: From Highly Polarized Sensory Neurons to Cancer Cells. Cells 2019, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Boyd, K.L.; Parekh, D.V.; Kehl-Fie, T.E.; Baldwin, H.S.; Brakebusch, C.; Skaar, E.P.; Boothby, M.; Zent, R. Cdc42 Promotes Host Defenses against Fatal Infection. Infect. Immun. 2013, 81, 2714–2723. [Google Scholar] [CrossRef] [Green Version]
- Gerasimcik, N.; Dahlberg, C.I.M.; Baptista, M.A.P.; Massaad, M.J.; Geha, R.S.; Westerberg, L.S.; Severinson, E. The Rho GTPase Cdc42 Is Essential for the Activation and Function of Mature B Cells. J. Immunol. 2015, 194, 4750–4758. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
| Clinical Phenotypes | Martinelli et al., 2018 [11] | Lam et al., 2019 [15] | Szczawinska-Poplonyk et al., 2020 [16] | Present Study | Total (%) |
|---|---|---|---|---|---|
| Intellectual disability | 11/15 | 1/4 | 1/1 | 0/1 | 13/21 (61.9%) |
| Seizures | 4/15 | 1/4 | 1/1 | 0/1 | 6/21 (28.5%) |
| Brain anomalies | 9/15 | 1/4 | 1/1 | 1/1 | 12/21 (57%) |
| Optic atrophy | 3/15 | 0/4 | 0/1 | 0/1 | 3/21 (14.3%) |
| Endocrine anomalies | 4/15 | 0/4 | 0/1 | 0/1 | 4/21 (19%) |
| Facial dysmorphism | 14/15 | 0/4 | 0/1 | 0/1 | 14/21 (66.7%) |
| Scoliosis/vertebral anomalies | 5/15 | 0/4 | 0/1 | 0/1 | 5/21 (23.8%) |
| Camptodacyly or other digit anomalies | 4/15 | 1/4 | 0/1 | 0/1 | 5/21 (23.8%) |
| Cardiac anomalies | 7/15 | 0/4 | 0/1 | 0/1 | 7/21 (33.3%) |
| Recurrent infections | 8/15 | 4/4 | 1/1 | 1/1 | 14/21 (66.7%) |
| Platelet anomalies (thrombocytopenia, macrothrombocytes | 5/15 | 4/4 | 1/1 | 1/1 | 11/21 (52.4%) |
| Skin rash | 0/15 | 4/4 | 1/1 | 1/1 | 6/21 (28.6%) |
| Hepatomegaly | 0/15 | 4/4 | 1/1 | 0/1 | 5/21 (23.8%) |
| Splenomegaly | 0/15 | 3/4 | 1/1 | 0/1 | 4/21 (19%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Asiri, A.; Alwadaani, D.; Umair, M.; Alhamoudi, K.M.; Almuhanna, M.H.; Nasir, A.; Alrfaei, B.M.; Al Tuwaijri, A.; Barhoumi, T.; Alyafee, Y.; et al. Pancytopenia, Recurrent Infection, Poor Wound Healing, Heterotopia of the Brain Probably Associated with A Candidate Novel de Novo CDC42 Gene Defect: Expanding the Molecular and Phenotypic Spectrum. Genes 2021, 12, 294. https://doi.org/10.3390/genes12020294
Asiri A, Alwadaani D, Umair M, Alhamoudi KM, Almuhanna MH, Nasir A, Alrfaei BM, Al Tuwaijri A, Barhoumi T, Alyafee Y, et al. Pancytopenia, Recurrent Infection, Poor Wound Healing, Heterotopia of the Brain Probably Associated with A Candidate Novel de Novo CDC42 Gene Defect: Expanding the Molecular and Phenotypic Spectrum. Genes. 2021; 12(2):294. https://doi.org/10.3390/genes12020294
Chicago/Turabian StyleAsiri, Abdulaziz, Deemah Alwadaani, Muhammad Umair, Kheloud M. Alhamoudi, Mohammed H. Almuhanna, Abdul Nasir, Bahauddeen M. Alrfaei, Abeer Al Tuwaijri, Tlili Barhoumi, Yusra Alyafee, and et al. 2021. "Pancytopenia, Recurrent Infection, Poor Wound Healing, Heterotopia of the Brain Probably Associated with A Candidate Novel de Novo CDC42 Gene Defect: Expanding the Molecular and Phenotypic Spectrum" Genes 12, no. 2: 294. https://doi.org/10.3390/genes12020294
APA StyleAsiri, A., Alwadaani, D., Umair, M., Alhamoudi, K. M., Almuhanna, M. H., Nasir, A., Alrfaei, B. M., Al Tuwaijri, A., Barhoumi, T., Alyafee, Y., Almuzzaini, B., Aldrees, M., Ballow, M., Alayyar, L., Al Abdulrahman, A., Alhaidan, Y., Al Ghasham, N., Al-Ajaji, S., Alsalamah, M., ... Alfadhel, M. (2021). Pancytopenia, Recurrent Infection, Poor Wound Healing, Heterotopia of the Brain Probably Associated with A Candidate Novel de Novo CDC42 Gene Defect: Expanding the Molecular and Phenotypic Spectrum. Genes, 12(2), 294. https://doi.org/10.3390/genes12020294