
Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data Acquisition, Filtering, and Manual Curation
2.2. Repeats Analyses and Genome Size Data
3. Results
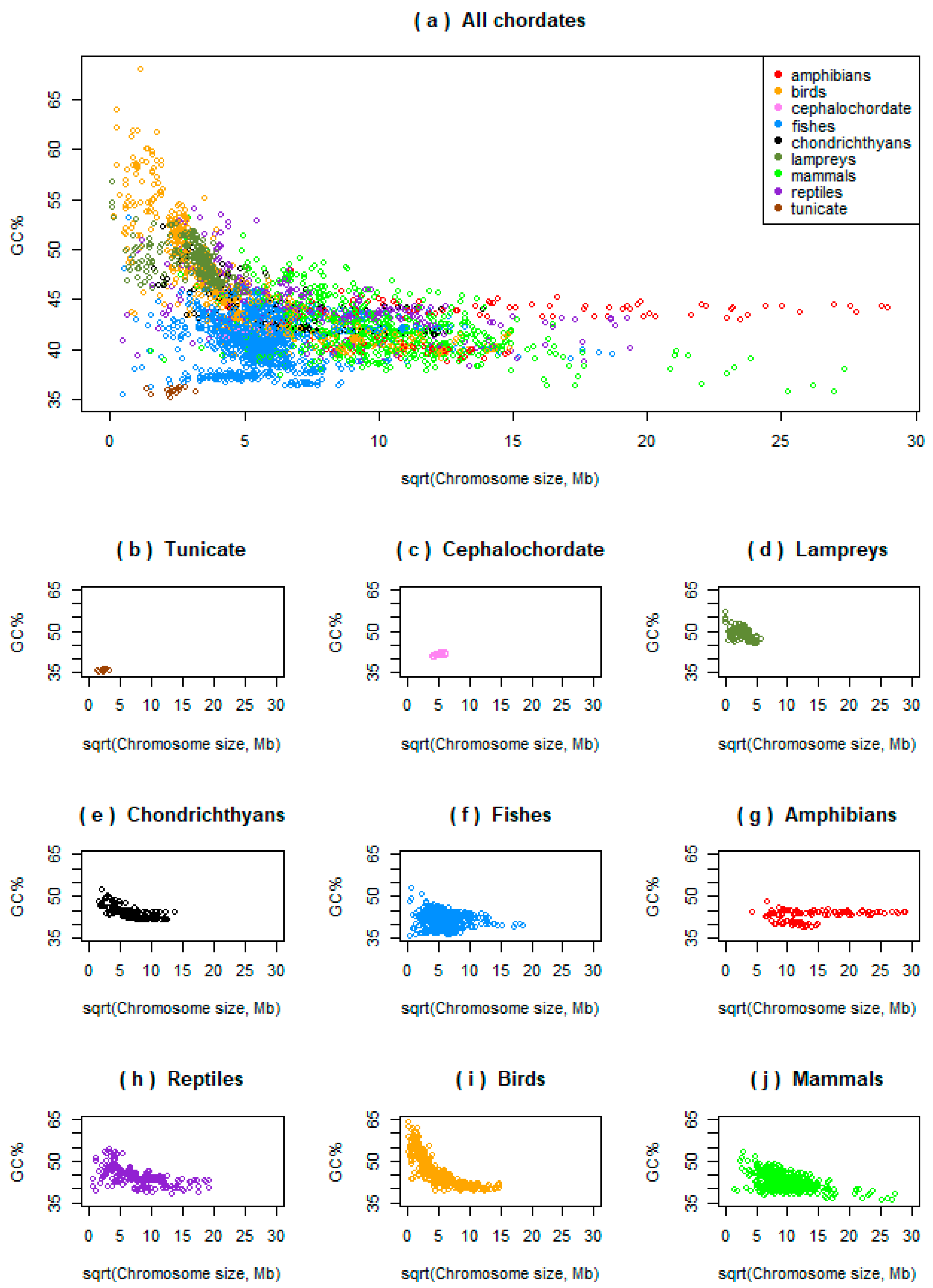
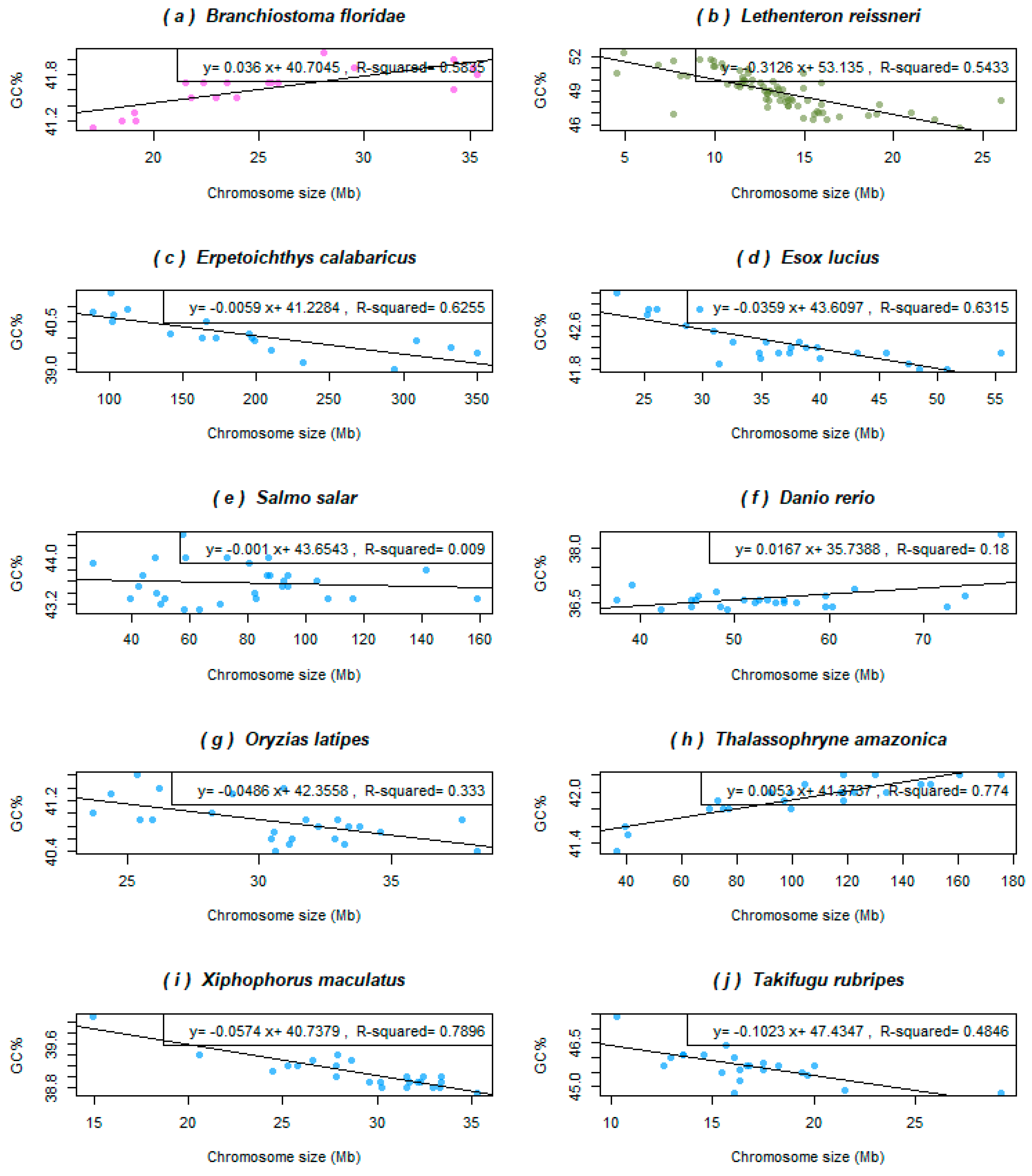
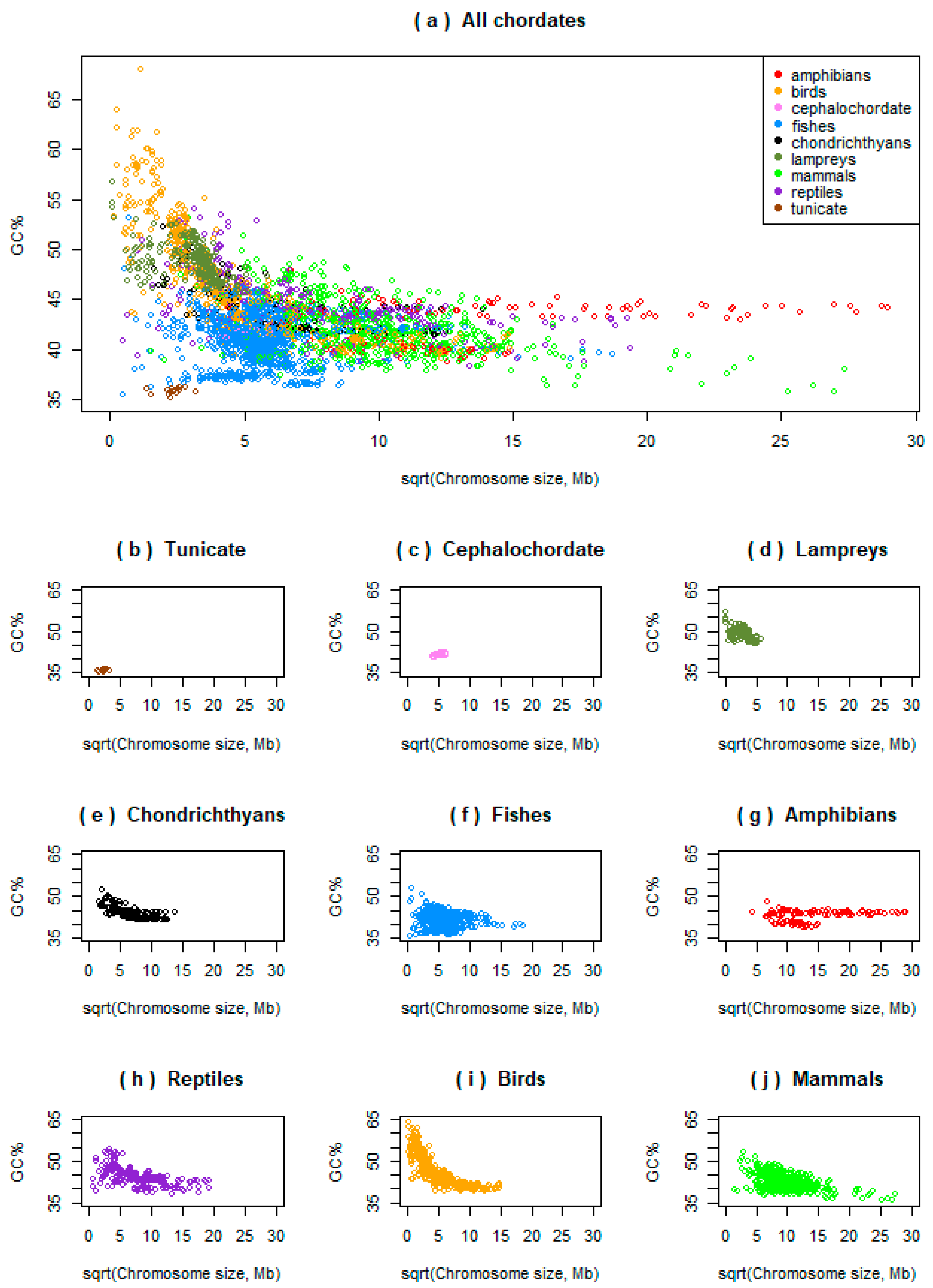
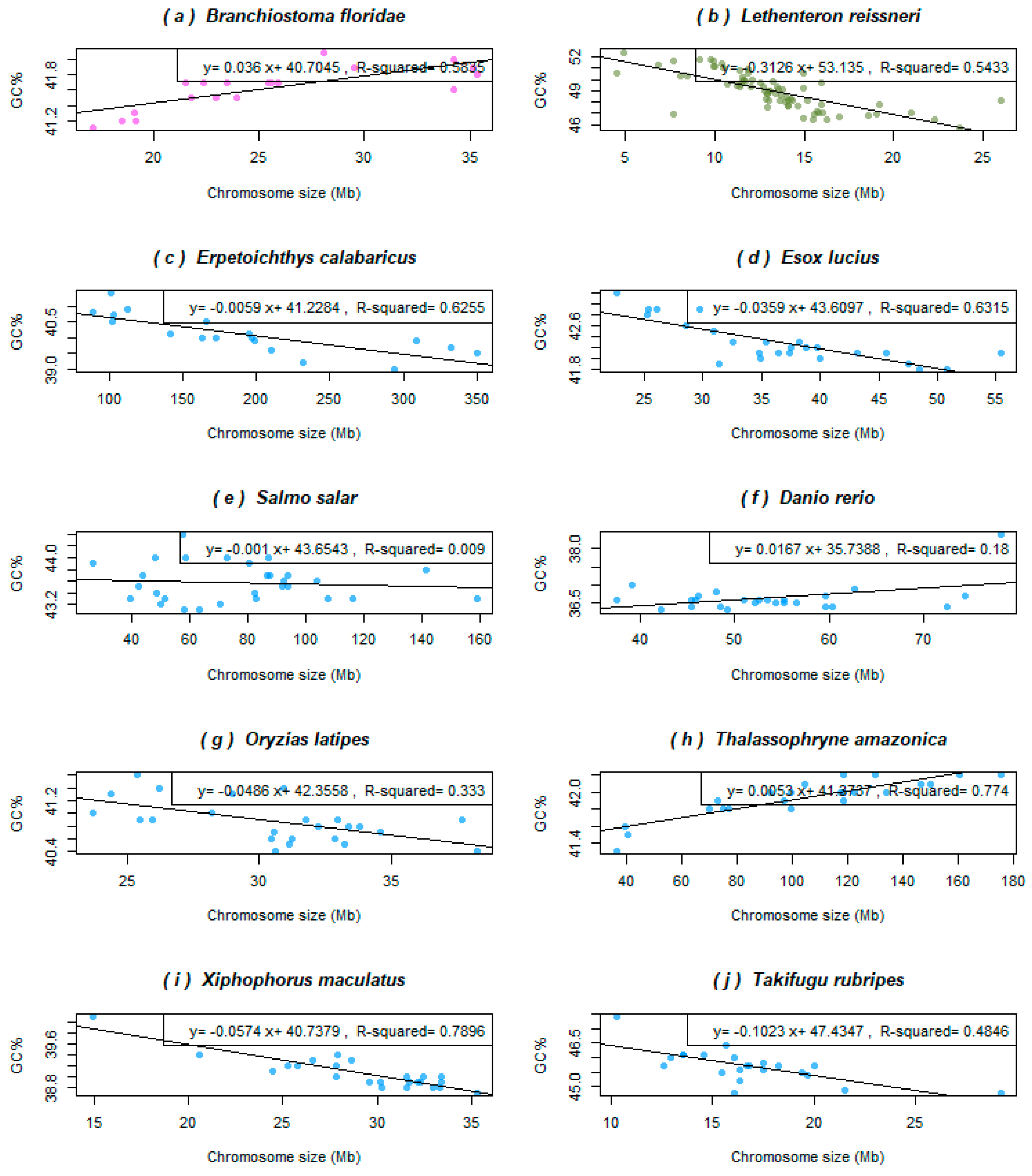
3.1. Variability in Relationships between the Chromosome Size and GC% in Ray-Finned Fishes
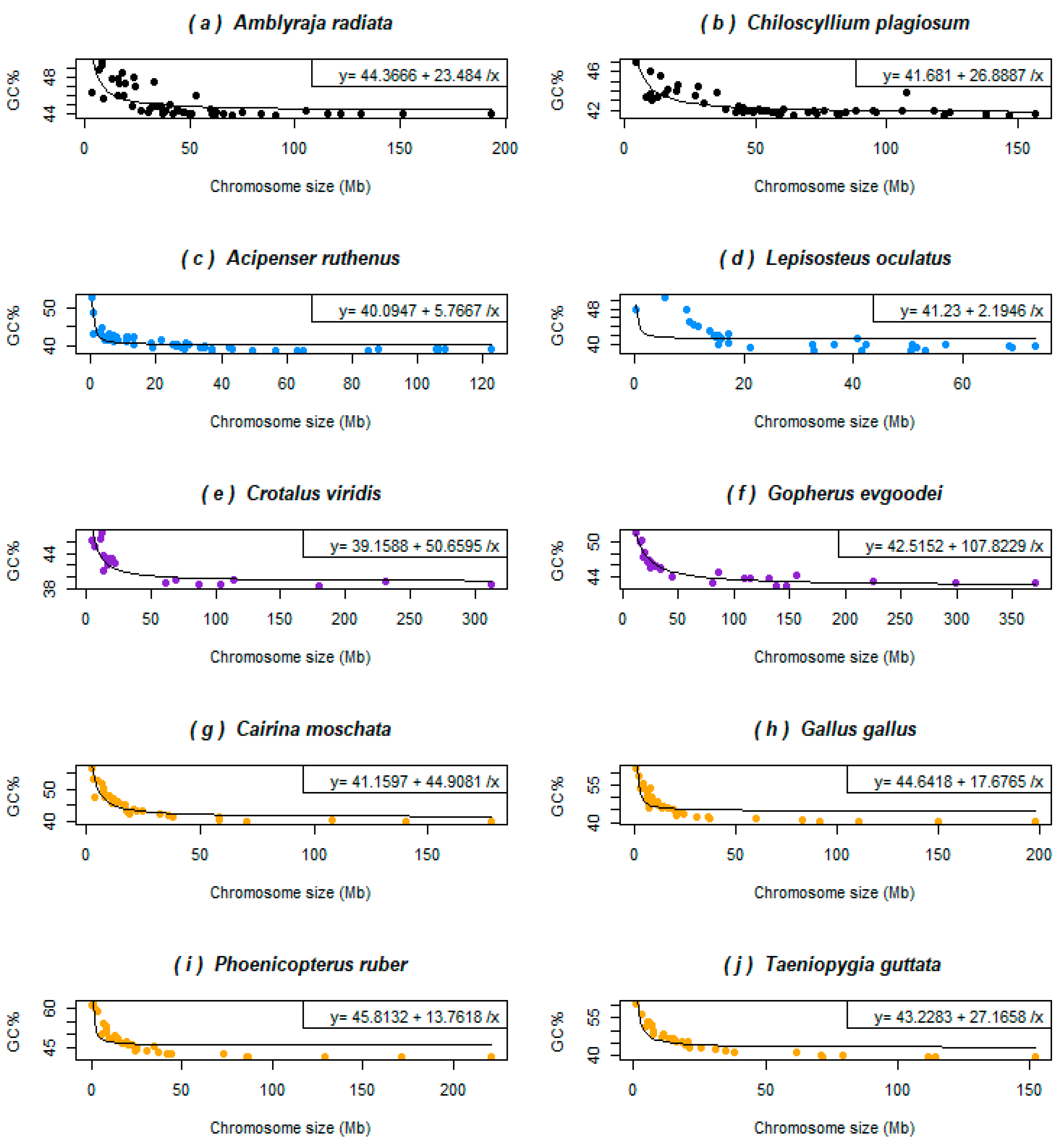
3.2. Basal Fish Lineages Show a Similar Relationship between the Chromosome Size and GC% to Birds and Some Reptiles
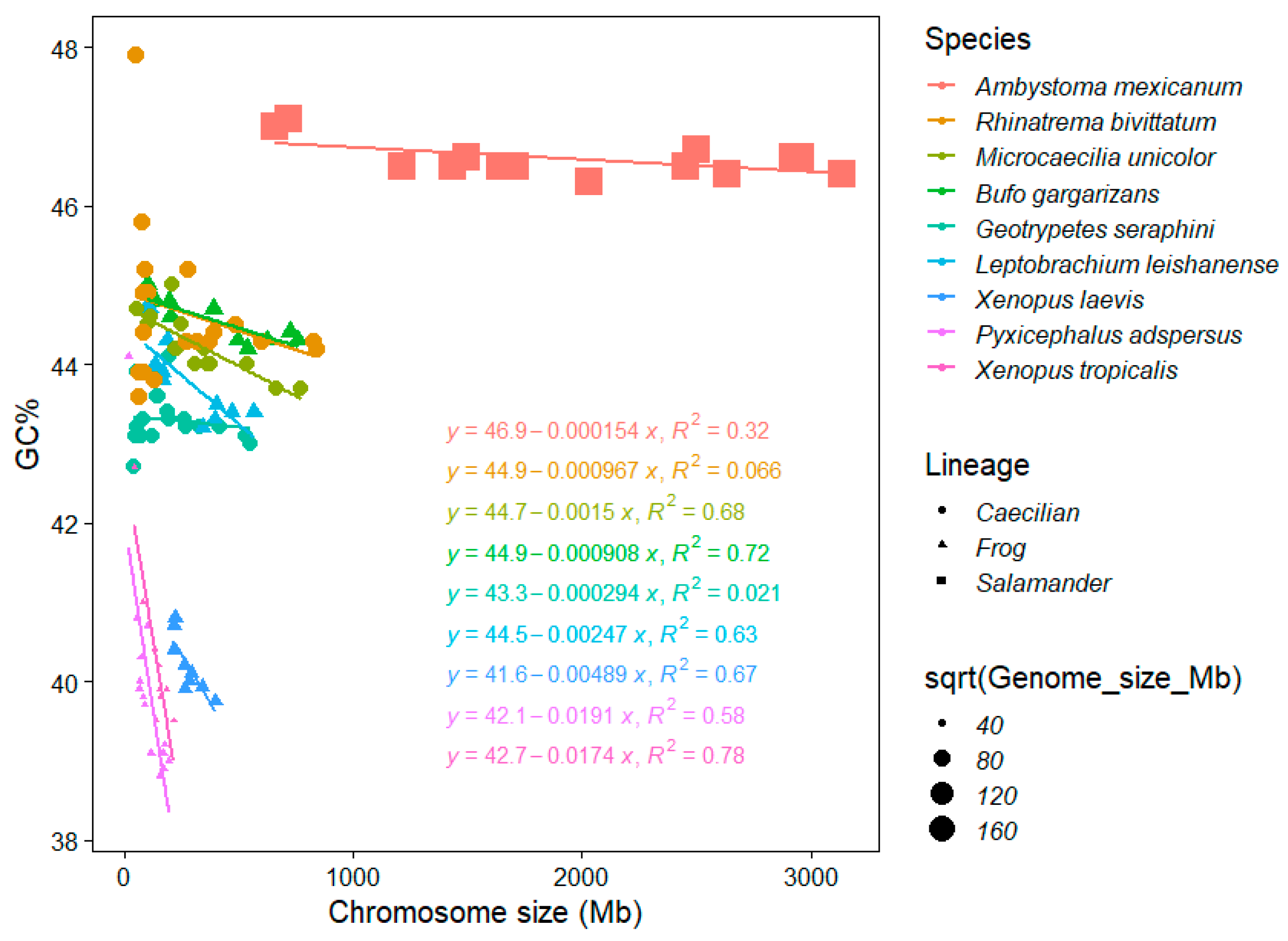
3.3. Genome Size Drives GC% in Amphibians but Inversely than Expected
- A single salamander species with its giant and extremely GC-rich chromosomes (and genome) showing a weak negative association between GC% and the chromosome size (R2 = 0.32; genome size 32,396.4 Mb, GC ~ 46.5%).
- Three caecilians and two frogs (pelobatid and bufonid) with an intermediate GC% and chromosome sizes (and intermediate genome sizes between 3779.43 and 5319.24 Mb, GC ~ 43%–44%) showing no association in two caecilians (R2 = 0.066 in Rhinatrema, R2 = 0.021 in Geotrypetes) to a significant negative association in the remaining caecilian Microcaecilia and two anurans (R2 = 0.63−0.72) between GC% and the chromosome size.
- Three remaining frogs (two pipids and a pyxicephalid) with a significantly (R2 = 0.5837, 0.6689, and 0.7824) negative association between GC% and the chromosome size (the smallest genomes between 1451.3 and 2718.43 Mb, GC ~ 39%–40.5%).
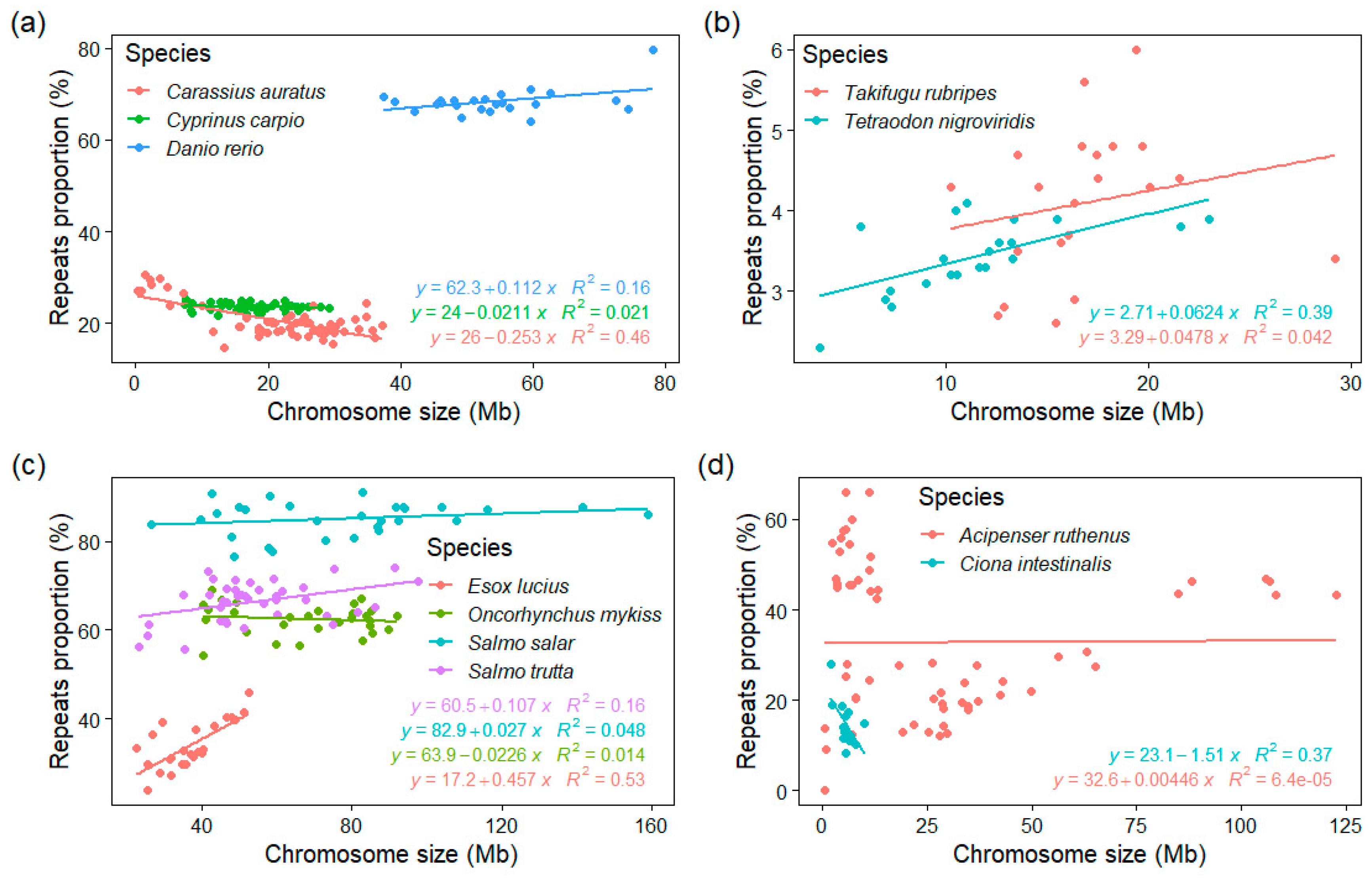
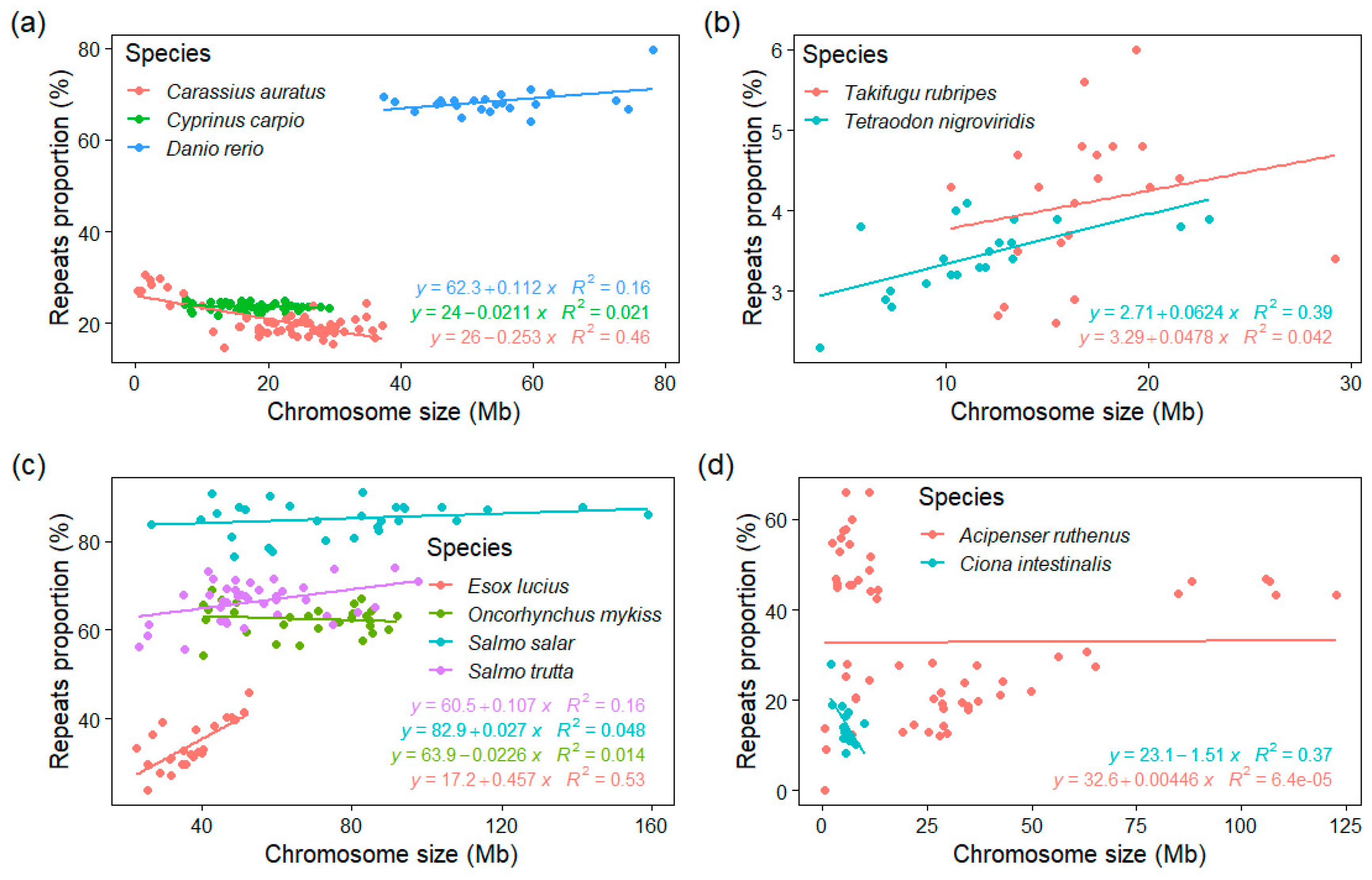
3.4. GC% vs. Repeats Proportion (%) and Chromosome Size
4. Discussion
4.1. Genome Size and Chromosome Numbers Do Not Entirely Explain the Difference in GC Evolution between Fish and Mammals
4.2. The Role of Chromosome Size Combines with the Influence of Repeats
4.3. Nucleotide Composition Investigations in Fish
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gregory, T.R. Animal Genome Size Database. Available online: http://www.genomesize.com (accessed on 31 January 2021).
- Levan, A.; Fredga, K.; Sandberg, A.A. Nomenclature for Centromeric Position on Chromosomes. Hereditas 1964, 52, 201–220. [Google Scholar] [CrossRef]
- Mark, H.F.; Mark, R.; Pan, T.; Mark, Y. Centromere Index Derivation by a Novel and Convenient Approach. Ann. Clin. Lab. Sci. 1993, 23, 267–274. [Google Scholar]
- Comings, D.E. Mechanisms of Chromosome Banding and Implications for Chromosome Structure. Annu. Rev. Genet. 1978, 12, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Luo, C. Multiple Chromosomal Banding in Grass Carp, Ctenopharyngodon Idellus. Heredity 1998, 81, 481–485. [Google Scholar] [CrossRef]
- Medrano, L.; Bernardi, G.; Couturier, J.; Dutrillaux, B.; Bernardi, G. Chromosome Banding and Genome Compartmentalization in Fishes. Chromosoma 1988, 96, 178–183. [Google Scholar] [CrossRef]
- Mayr, B.; Kalat, M.; Ráb, P.; Lambrou, M. Band Karyotypes and Specific Types of Heterochromatins in Several Species of European Percid Fishes (Percidea, Pisces). Genetica 1987, 75, 199–205. [Google Scholar] [CrossRef]
- Mank, J.E.; Avise, J.C. Phylogenetic Conservation of Chromosome Numbers in Actinopterygiian Fishes. Genetica 2006, 127, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Gregory, T.R.; Witt, J.D.S. Population Size and Genome Size in Fishes: A Closer Look. Genome 2008, 51, 309–313. [Google Scholar] [CrossRef]
- Hardie, D.C.; Hebert, P.D.N. The Nucleotypic Effects of Cellular DNA Content in Cartilaginous and Ray-Finned Fishes. Genome 2003, 46, 683–706. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.C.; Hebert, P.D. Genome-Size Evolution in Fishes. Can. J. Fish. Aquat. Sci. 2004, 61, 1636–1646. [Google Scholar] [CrossRef]
- Melodelima, C.; Gautier, C. The GC-Heterogeneity of Teleost Fishes. BMC Genom. 2008, 9, 632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costantini, M.; Auletta, F.; Bernardi, G. Isochore Patterns and Gene Distributions in Fish Genomes. Genomics 2007, 90, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Costantini, M.; Cammarano, R.; Bernardi, G. The Evolution of Isochore Patterns in Vertebrate Genomes. BMC Genom. 2009, 10, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eyre-Walker, A. Recombination and Mammalian Genome Evolution. Proc. R. Soc. Lond. B Biol. Sci. 1993, 252, 237–243. [Google Scholar] [CrossRef]
- Fullerton, S.M.; Bernardo Carvalho, A.; Clark, A.G. Local Rates of Recombination Are Positively Correlated with GC Content in the Human Genome. Mol. Biol. Evol. 2001, 18, 1139–1142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montoya-Burgos, J.I.; Boursot, P.; Galtier, N. Recombination Explains Isochores in Mammalian Genomes. Trends Genet. 2003, 19, 128–130. [Google Scholar] [CrossRef]
- Mugal, C.F.; Weber, C.C.; Ellegren, H. GC-Biased Gene Conversion Links the Recombination Landscape and Demography to Genomic Base Composition: GC-Biased Gene Conversion Drives Genomic Base Composition across a Wide Range of Species. BioEssays 2015, 37, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Stapley, J.; Feulner, P.G.D.; Johnston, S.E.; Santure, A.W.; Smadja, C.M. Variation in Recombination Frequency and Distribution across Eukaryotes: Patterns and Processes. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160455. [Google Scholar] [CrossRef]
- Romiguier, J.; Ranwez, V.; Douzery, E.J.P.; Galtier, N. Contrasting GC-Content Dynamics across 33 Mammalian Genomes: Relationship with Life-History Traits and Chromosome Sizes. Genome Res. 2010, 20, 1001–1009. [Google Scholar] [CrossRef] [Green Version]
- Huttener, R.; Thorrez, L.; In’t Veld, T.; Granvik, M.; Snoeck, L.; Van Lommel, L.; Schuit, F. GC Content of Vertebrate Exome Landscapes Reveal Areas of Accelerated Protein Evolution. BMC Evol. Biol. 2019, 19. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, D.; Ye, B.; Shi, M.; Ma, L.; Zhang, Y.; Zhao, Z. GC3-Biased Gene Domains in Mammalian Genomes. Bioinformatics 2015, 31, 3081–3084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C.C.; Boussau, B.; Romiguier, J.; Jarvis, E.D.; Ellegren, H. Evidence for GC-Biased Gene Conversion as a Driver of between-Lineage Differences in Avian Base Composition. Genome Biol. 2014, 15. [Google Scholar] [CrossRef]
- Bolívar, P.; Mugal, C.F.; Nater, A.; Ellegren, H. Recombination Rate Variation Modulates Gene Sequence Evolution Mainly via GC-Biased Gene Conversion, Not Hill–Robertson Interference, in an Avian System. Mol. Biol. Evol. 2016, 33, 216–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsubara, K.; Kuraku, S.; Tarui, H.; Nishimura, O.; Nishida, C.; Agata, K.; Kumazawa, Y.; Matsuda, Y. Intra-Genomic GC Heterogeneity in Sauropsids: Evolutionary Insights from CDNA Mapping and GC3 Profiling in Snake. BMC Genom. 2012, 13, 604. [Google Scholar] [CrossRef] [Green Version]
- Figuet, E.; Ballenghien, M.; Romiguier, J.; Galtier, N. Biased Gene Conversion and GC-Content Evolution in the Coding Sequences of Reptiles and Vertebrates. Genome Biol. Evol. 2015, 7, 240–250. [Google Scholar] [CrossRef] [Green Version]
- Wang, D. GCevobase: An Evolution-Based Database for GC Content in Eukaryotic Genomes. Bioinformatics 2018, 34, 2129–2131. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, S.; Kirzhner, V.; Korol, A. Organizational Heterogeneity of Vertebrate Genomes. PLoS ONE 2012, 7, e32076. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-Q.; Du, D. Variation, Evolution, and Correlation Analysis of C+G Content and Genome or Chromosome Size in Different Kingdoms and Phyla. PLoS ONE 2014, 9, e88339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symonová, R.; Suh, A. Nucleotide Composition of Transposable Elements Likely Contributes to AT/GC Compositional Homogeneity of Teleost Fish Genomes. Mob. DNA 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Carducci, F.; Barucca, M.; Canapa, A.; Carotti, E.; Biscotti, M.A. Mobile Elements in Ray-Finned Fish Genomes. Life 2020, 10, 221. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. The Vertebrate Genome: Isochores and Evolution. Mol. Biol. Evol. 1993. [Google Scholar] [CrossRef] [Green Version]
- Symonová, R.; Majtánová, Z.; Arias-Rodriguez, L.; Mořkovský, L.; Kořínková, T.; Cavin, L.; Pokorná, M.J.; Doležálková, M.; Flajšhans, M.; Normandeau, E.; et al. Genome Compositional Organization in Gars Shows More Similarities to Mammals than to Other Ray-Finned Fish: Cytogenomics of Gars. J. Exp. Zool. Part B Mol. Dev. Evol. 2017, 328, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Matoulek, D.; Borůvková, V.; Ocalewicz, K.; Symonová, R. GC and Repeats Profiling along Chromosomes—The Future of Fish Compositional Cytogenomics. Genes 2021, 12, 50. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, G. Structural and Evolutionary Genomics Natural Selection in Genome Evolution; Elsevier: Amsterdam, The Netherlands, 2005. [Google Scholar]
- Peona, V.; Weissensteiner, M.H.; Suh, A. How Complete Are “Complete” Genome Assemblies?—An Avian Perspective. Mol. Ecol. Resour. 2018, 18, 1188–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jebb, D.; Huang, Z.; Pippel, M.; Hughes, G.M.; Lavrichenko, K.; Devanna, P.; Winkler, S.; Jermiin, L.S.; Skirmuntt, E.C.; Katzourakis, A.; et al. Six Reference-Quality Genomes Reveal Evolution of Bat Adaptations. Nature 2020, 583, 578–584. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; Version 2.6.2; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Use R! Springer International Publishing: Cham, Switzerland, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Nelson, J.S.; Grande, T.; Wilson, M.V.H. Fishes of the World, 5th ed.; John Wiley & Sons: Hoboken, NJ, USA, 2016; ISBN 978-1-118-34233-6. [Google Scholar]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The Zebrafish Reference Genome Sequence and Its Relationship to the Human Genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hidalgo, O.; Pellicer, J.; Christenhusz, M.; Schneider, H.; Leitch, A.R.; Leitch, I.J. Is There an Upper Limit to Genome Size? Trends Plant Sci. 2017, 22, 567–573. [Google Scholar] [CrossRef] [PubMed]
- NCBI Genome Browser. Available online: https://www.ncbi.nlm.nih.gov/genome/browse (accessed on 31 January 2021).
- Meyer, A.; Schartl, M. Gene and Genome Duplications in Vertebrates: The One-to-Four (-to-Eight in Fish) Rule and the Evolution of Novel Gene Functions. Curr. Opin. Cell Biol. 1999, 11, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Hannan, A.J. Tandem Repeats and Repeatomes: Delving Deeper into the ‘Dark Matter’ of Genomes. EBioMedicine 2018, 31, 3–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arkhipova, I.R.; Yushenova, I.A. Giant Transposons in Eukaryotes: Is Bigger Better? Genome Biol. Evol. 2019, 11, 906–918. [Google Scholar] [CrossRef] [Green Version]
- Fontana, F.; Bruch, R.M.; Binkowski, F.P.; Lanfredi, M.; Chicca, M.; Beltrami, N.; Congiu, L. Karyotype Characterization of the Lake Sturgeon, Acipenser fulvescens (Rafinesque 1817) by Chromosome Banding and Fluorescent in Situ Hybridization. Genome 2004, 47, 742–746. [Google Scholar] [CrossRef]
- Symonová, R.; Havelka, M.; Amemiya, C.T.; Howell, W.M.; Kořínková, T.; Flajšhans, M.; Gela, D.; Ráb, P. Molecular Cytogenetic Differentiation of Paralogs of Hox Paralogs in Duplicated and Re-Diploidized Genome of the North American Paddlefish (Polyodon spathula). BMC Genet. 2017, 18. [Google Scholar] [CrossRef] [Green Version]
- Majtánová, Z.; Symonová, R.; Arias-Rodriguez, L.; Sallan, L.; Ráb, P. “Holostei versus Halecostomi” Problem: Insight from Cytogenetics of Ancient Nonteleost Actinopterygian Fish, Bowfin Amia calva: Molecular Cytogenetics of Amia calva. J. Exp. Zool. Part B Mol. Dev. Evol. 2017, 328, 620–628. [Google Scholar] [CrossRef]
- Gaffaroglu, M.; Majtánová, Z.; Symonová, R.; Pelikánová, Š.; Unal, S.; Lajbner, Z.; Ráb, P. Present and Future Salmonid Cytogenetics. Genes 2020, 11, 1462. [Google Scholar] [CrossRef]
- Lien, S.; Koop, B.F.; Sandve, S.R.; Miller, J.R.; Kent, M.P.; Nome, T.; Hvidsten, T.R.; Leong, J.S.; Minkley, D.R.; Zimin, A.; et al. The Atlantic Salmon Genome Provides Insights into Rediploidization. Nature 2016, 533, 200–205. [Google Scholar] [CrossRef] [Green Version]
- De-Kayne, R.; Zoller, S.; Feulner, P.G.D. A de Novo Chromosome-level Genome Assembly of Coregonus sp. “Balchen”: One Representative of the Swiss Alpine Whitefish Radiation. Mol. Ecol. Resour. 2020, 20, 1093–1109. [Google Scholar] [CrossRef]
- Pearse, D.E.; Barson, N.J.; Nome, T.; Gao, G.; Campbell, M.A.; Abadía-Cardoso, A.; Anderson, E.C.; Rundio, D.E.; Williams, T.H.; Naish, K.A.; et al. Sex-Dependent Dominance Maintains Migration Supergene in Rainbow Trout. Nat. Ecol. Evol. 2019, 3, 1731–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christensen, K.A.; Leong, J.S.; Sakhrani, D.; Biagi, C.A.; Minkley, D.R.; Withler, R.E.; Rondeau, E.B.; Koop, B.F.; Devlin, R.H. Chinook Salmon (Oncorhynchus tshawytscha) Genome and Transcriptome. PLoS ONE 2018, 13, e0195461. [Google Scholar] [CrossRef] [Green Version]
- Canapa, A.; Barucca, M.; Biscotti, M.A.; Forconi, M.; Olmo, E. Transposons, Genome Size, and Evolutionary Insights in Animals. Cytogenet. Genome Res. 2015, 147, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Nowoshilow, S.; Schloissnig, S.; Fei, J.-F.; Dahl, A.; Pang, A.W.C.; Pippel, M.; Winkler, S.; Hastie, A.R.; Young, G.; Roscito, J.G.; et al. The Axolotl Genome and the Evolution of Key Tissue Formation Regulators. Nature 2018, 554, 50–55. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, F.; Arkhipova, I.R. Transposable Elements and Polyploid Evolution in Animals. Curr. Opin. Genet. Dev. 2018, 49, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, A.; Angelini, C.; Sanges, R.; Yagi, M.; Agnisola, C.; D’Onofrio, G. On the Genome Base Composition of Teleosts: The Effect of Environment and Lifestyle. BMC Genom. 2016, 17. [Google Scholar] [CrossRef] [Green Version]
- Yi, S.; Streelman, J.T. Genome Size Is Negatively Correlated with Effective Population Size in Ray-Finned Fish. Trends Genet. 2005, 21, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Rolland, J.; Schluter, D.; Romiguier, J. Vulnerability to Fishing and Life History Traits Correlate with the Load of Deleterious Mutations in Teleosts. Mol. Biol. Evol. 2020, 37, 2192–2196. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boissinot, S. The Evolutionary Dynamics of Transposable Elements in Eukaryote Genomes. In Genome Dynamics; Garrido-Ramos, M.A., Ed.; S. KARGER AG: Basel, Switzerland, 2012; Volume 7, pp. 68–91. ISBN 978-3-318-02149-3. [Google Scholar]
- Bourgeois, Y.; Boissinot, S. On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes 2019, 10, 419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, T.V.; Uzunović, J.; Wright, S.I. Coevolution between Transposable Elements and Recombination. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160458. [Google Scholar] [CrossRef]
- Ruggiero, R.P.; Boissinot, S. Variation in Base Composition Underlies Functional and Evolutionary Divergence in Non-LTR Retrotransposons. Mob. DNA 2020, 11. [Google Scholar] [CrossRef]
- Paudel, R.; Fedorova, L.; Fedorov, A. Adapting Biased Gene Conversion Theory to Account for Intensive GC-Content Deterioration in the Human Genome by Novel Mutations. PLoS ONE 2020, 15, e0232167. [Google Scholar] [CrossRef]
- Nam, K.; Ellegren, H. Recombination Drives Vertebrate Genome Contraction. PLoS Genet. 2012, 8, e1002680. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borůvková, V.; Howell, W.M.; Matoulek, D.; Symonová, R. Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution. Genes 2021, 12, 312. https://doi.org/10.3390/genes12020312
Borůvková V, Howell WM, Matoulek D, Symonová R. Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution. Genes. 2021; 12(2):312. https://doi.org/10.3390/genes12020312
Chicago/Turabian StyleBorůvková, Veronika, W. Mike Howell, Dominik Matoulek, and Radka Symonová. 2021. "Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution" Genes 12, no. 2: 312. https://doi.org/10.3390/genes12020312
APA StyleBorůvková, V., Howell, W. M., Matoulek, D., & Symonová, R. (2021). Quantitative Approach to Fish Cytogenetics in the Context of Vertebrate Genome Evolution. Genes, 12(2), 312. https://doi.org/10.3390/genes12020312