
DNN-m6A: A Cross-Species Method for Identifying RNA N6-methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion



Abstract
1. Introduction
2. Materials and Methods
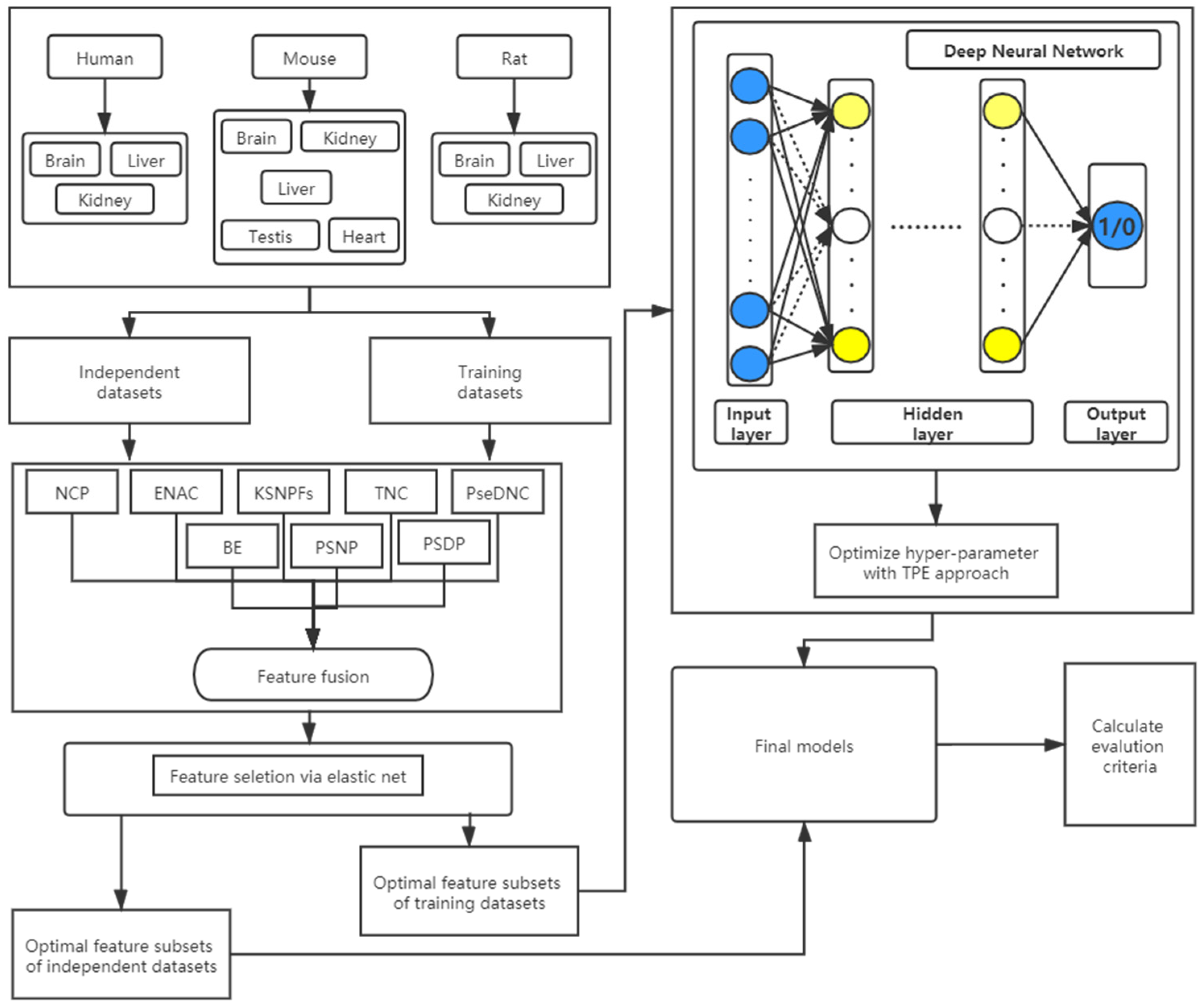
2.1. Benchmark Datasets
2.2. Feature Extraction
2.2.1. Binary Encoding (BE)
2.2.2. Nucleotide Composition (NC)
2.2.3. K-Spaced Nucleotide Pair Frequencies (KSNPFs)
2.2.4. Position-Specific Nucleotide Propensity (PSNP) and Position-Specific Dinucleotide Propensity (PSDP)
2.2.5. Nucleotide Chemical Property (NCP)
2.2.6. Pseudo Dinucleotide Composition (PseDNC)
2.3. Feature Selection Method
2.4. Deep Neural Network
2.5. Hyper-Parameter Optimization
2.6. Performance Evolution
2.7. Description of the DNN-m6A Process
3. Results and Discussion
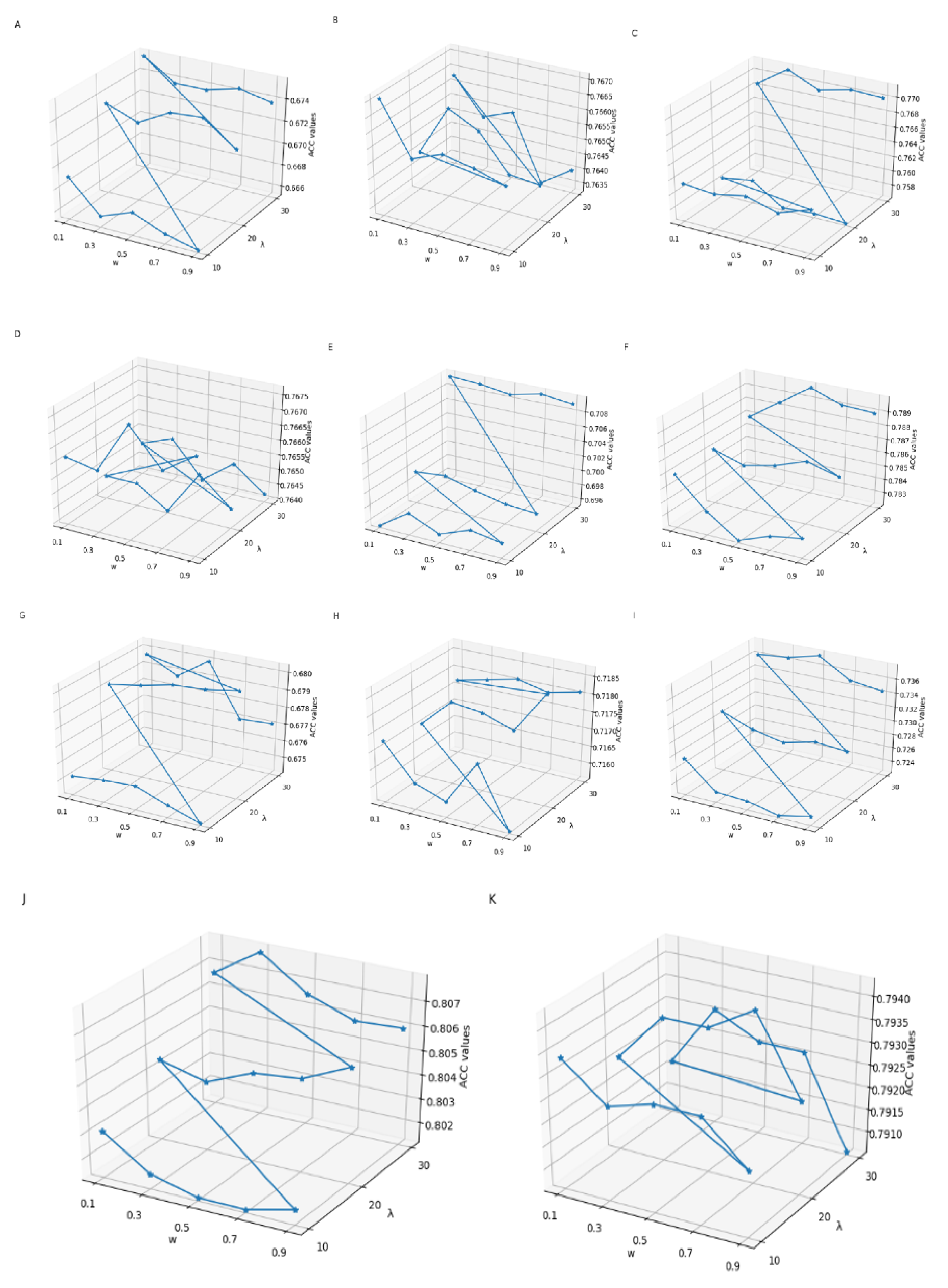
3.1. Parameter Selection of Feature Extraction
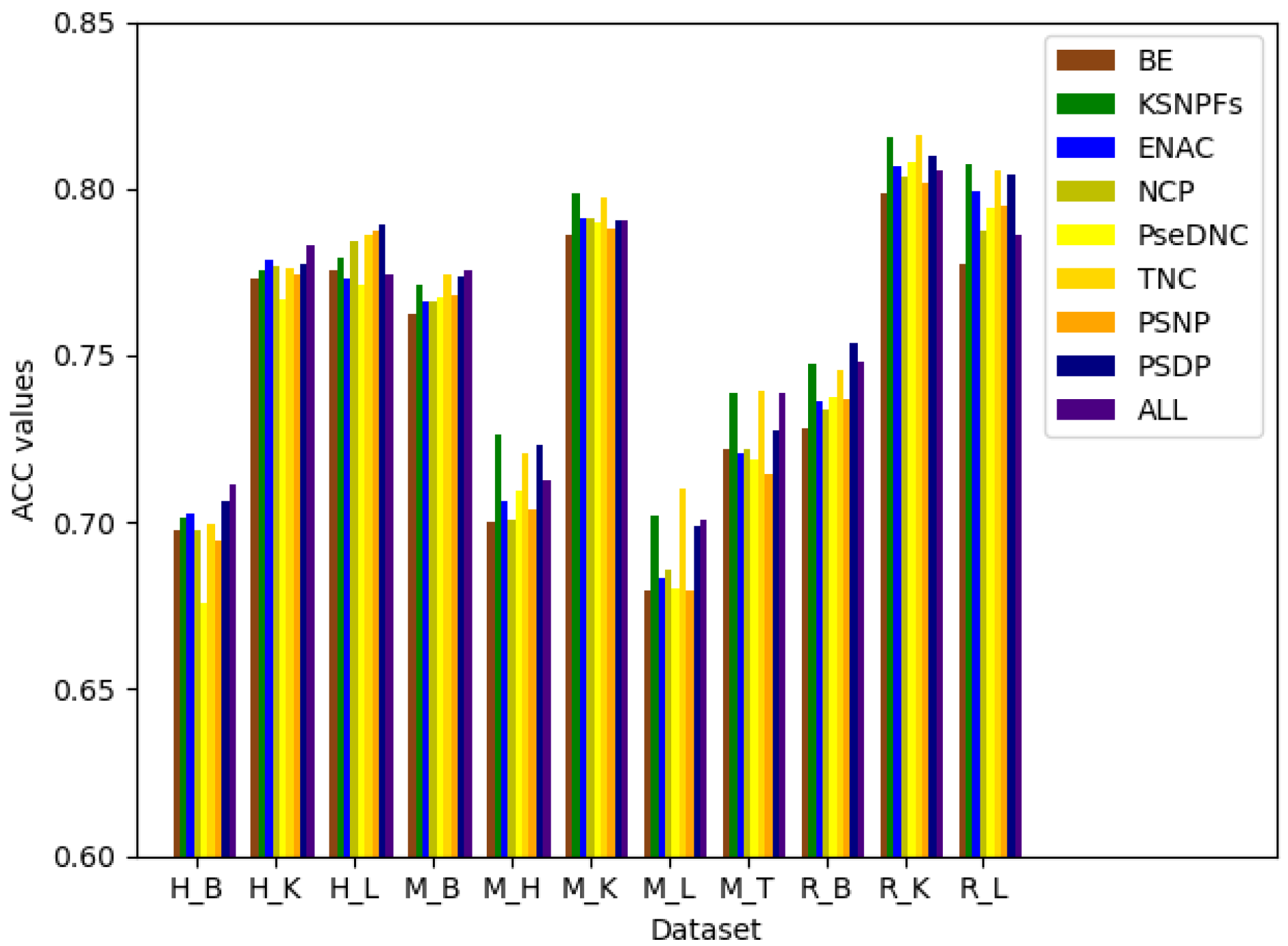
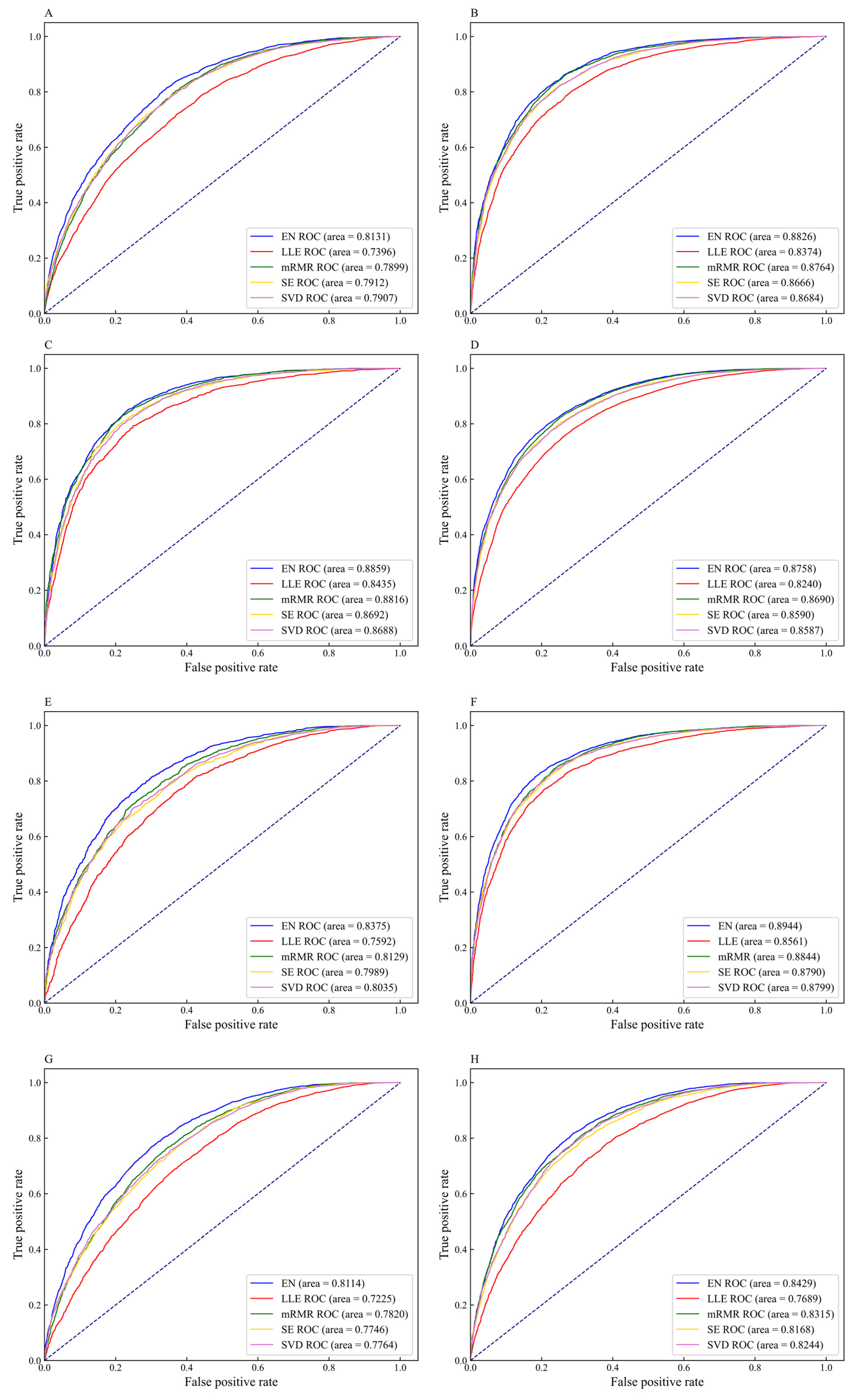
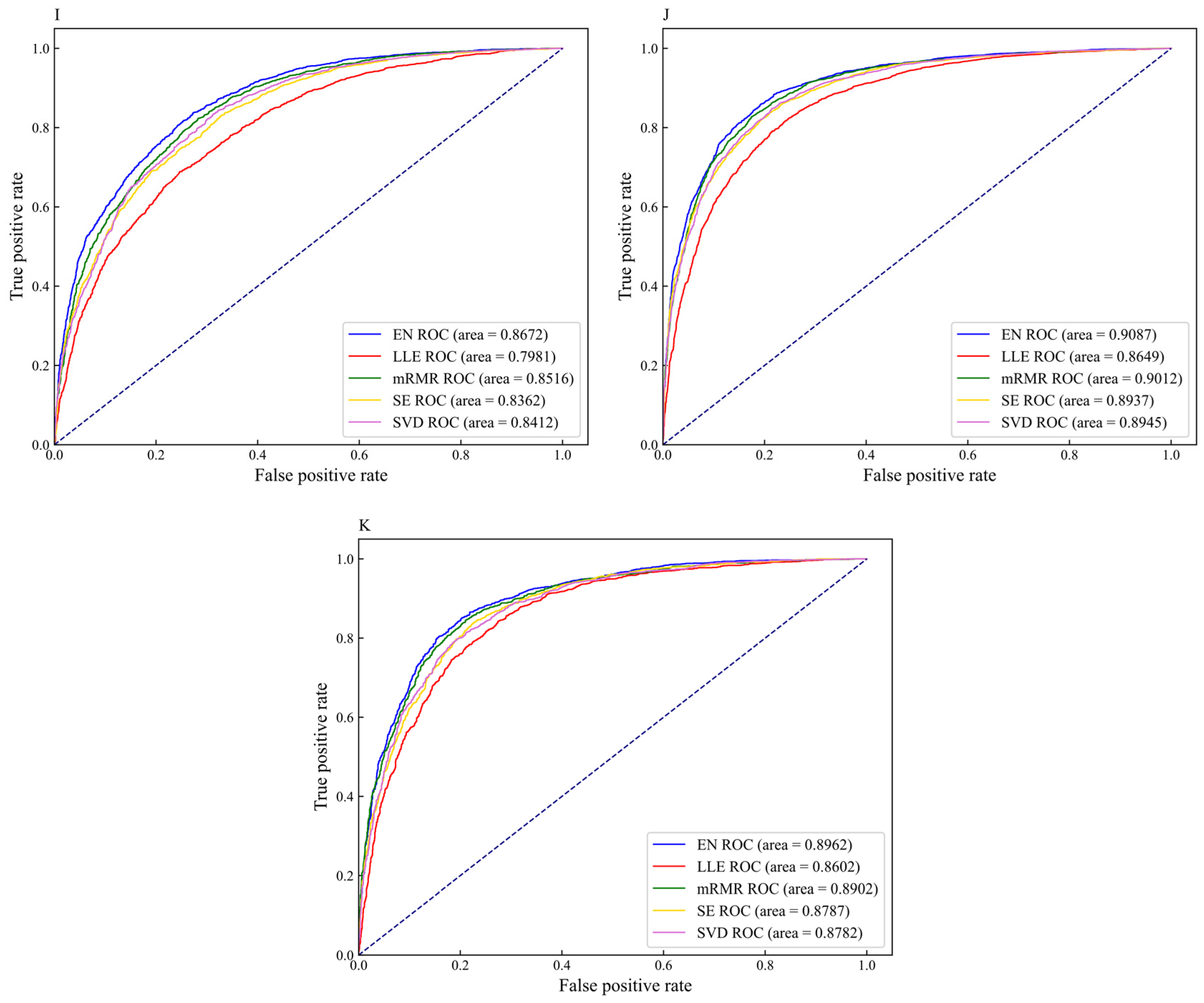
3.2. The Performance of Feature Extraction Methods
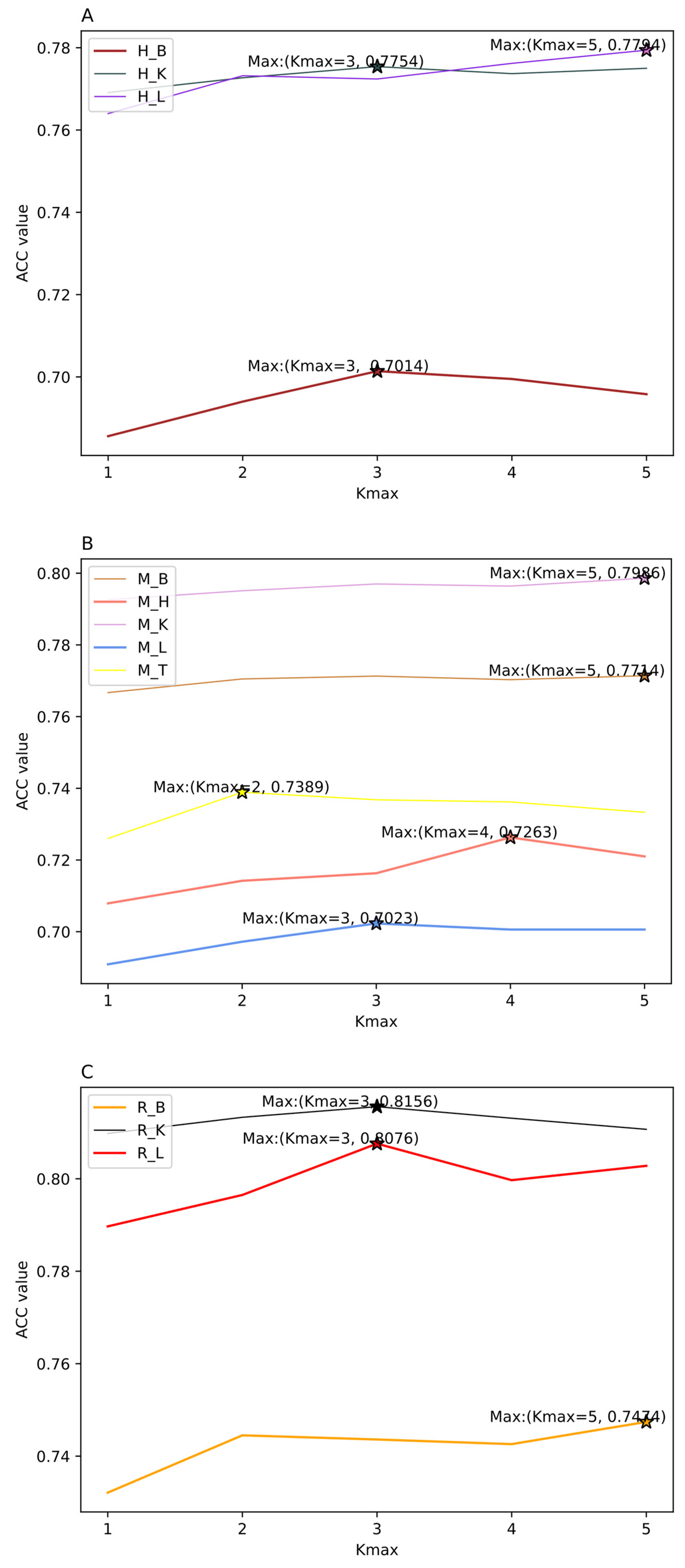
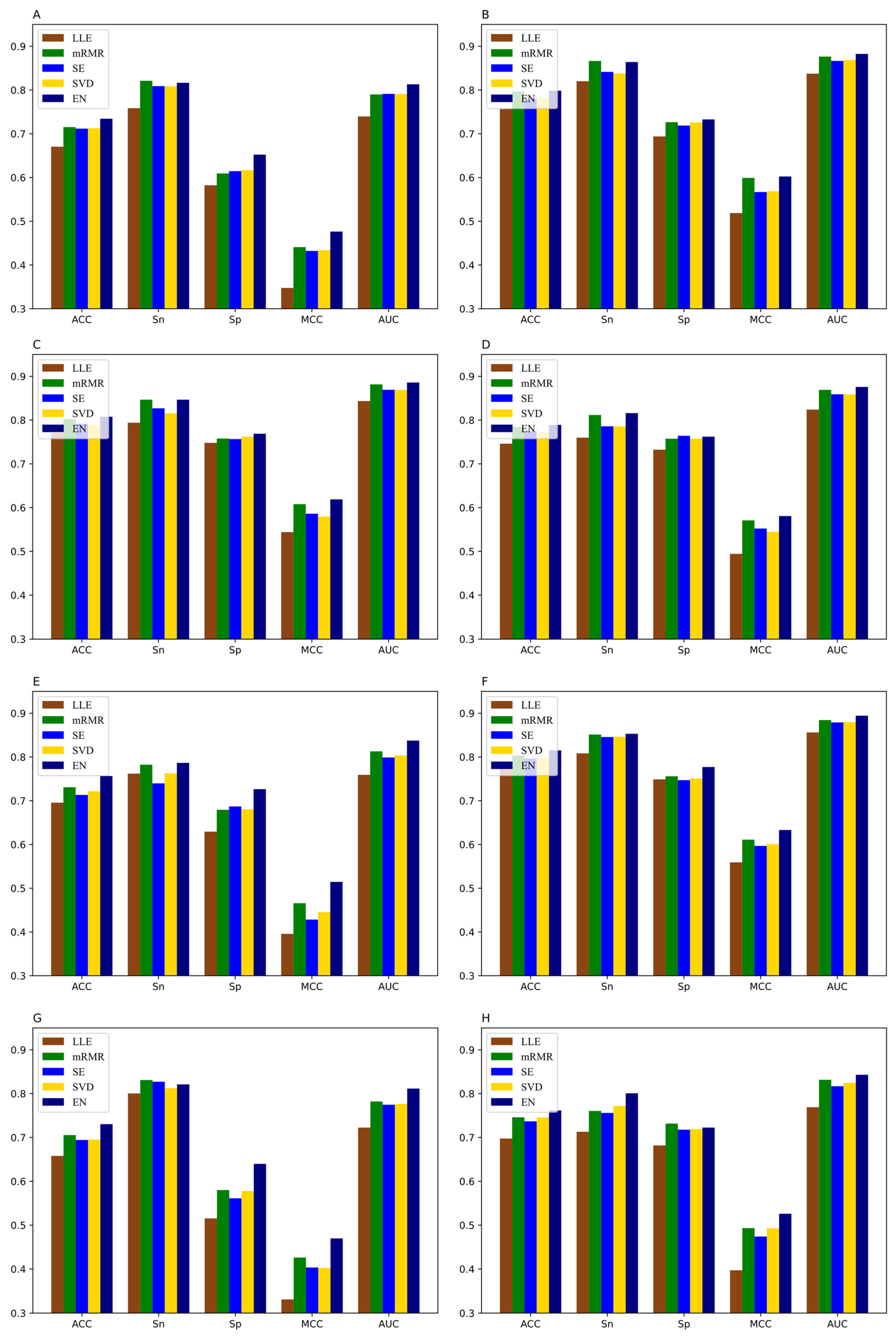
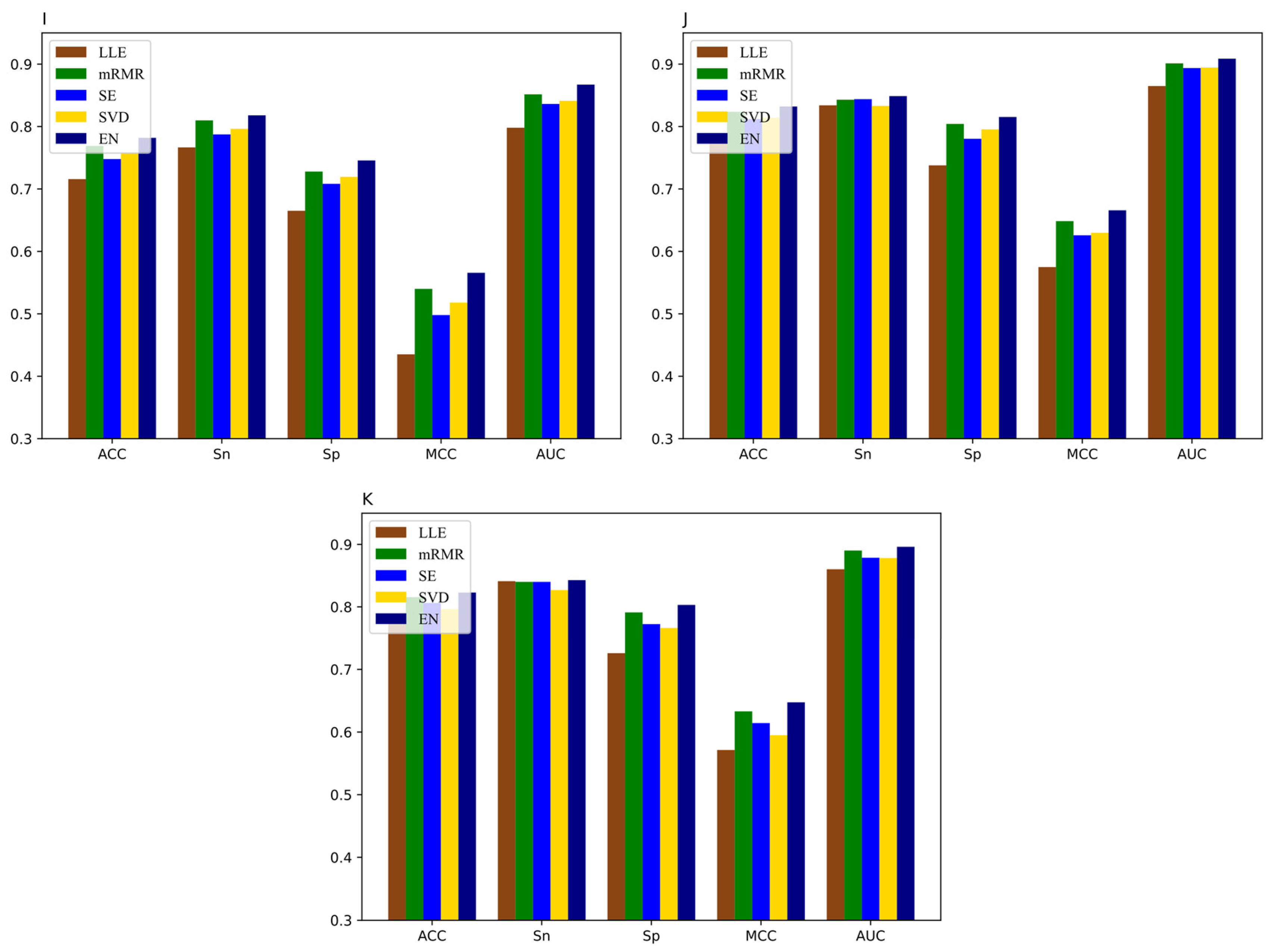
3.3. The Performance of Feature Selection Methods
3.4. The Performance of Hyper-Parameter Optimization
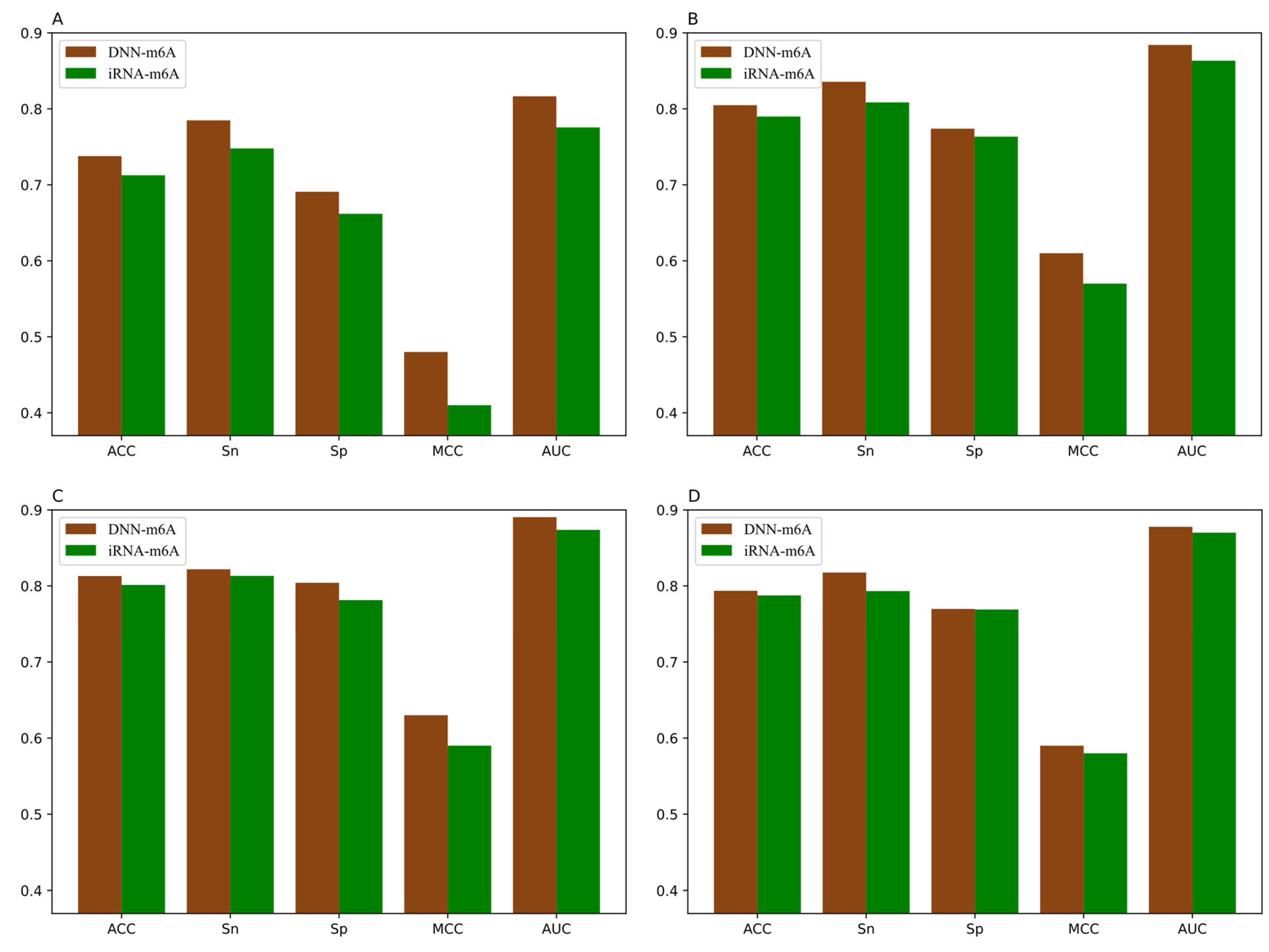
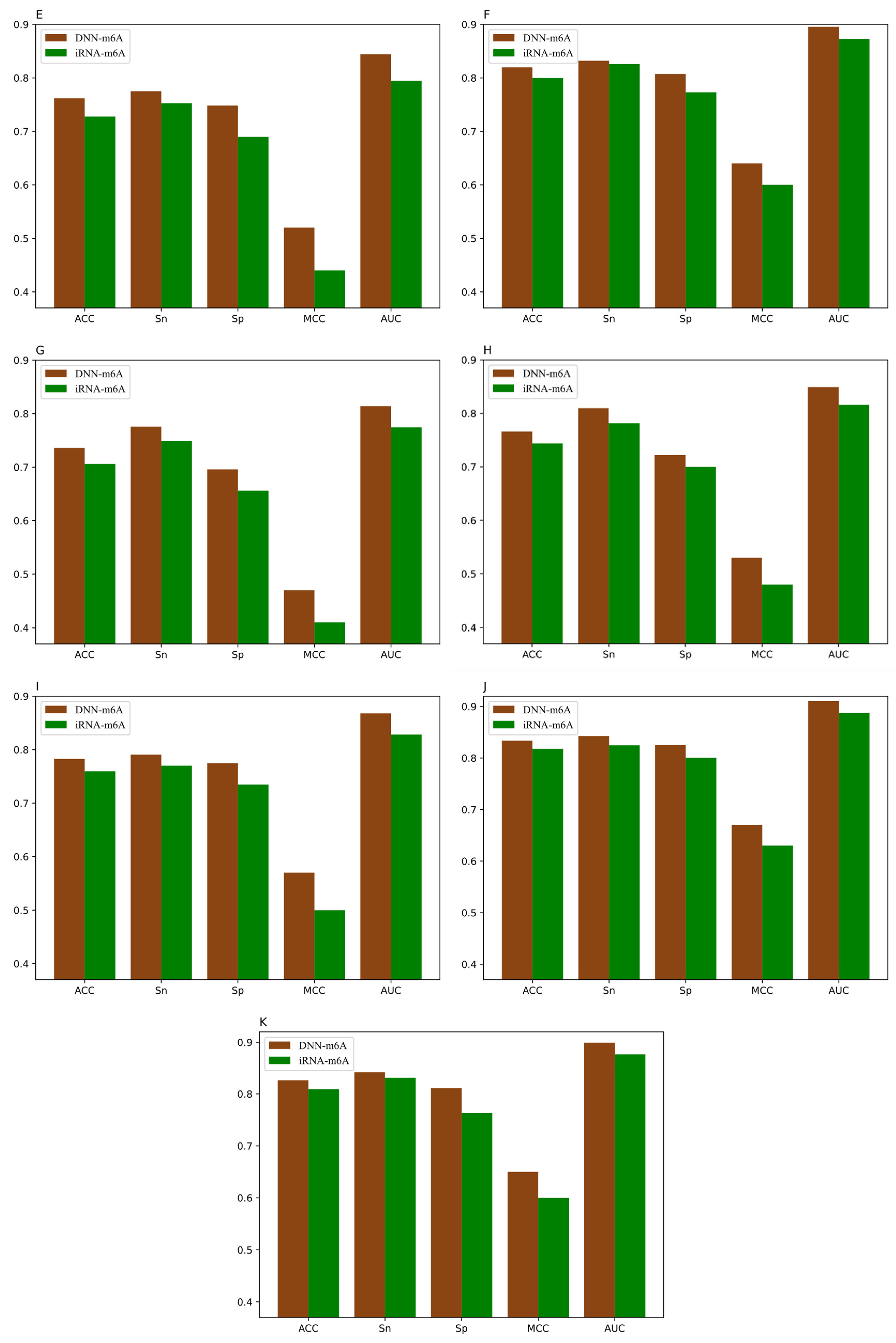
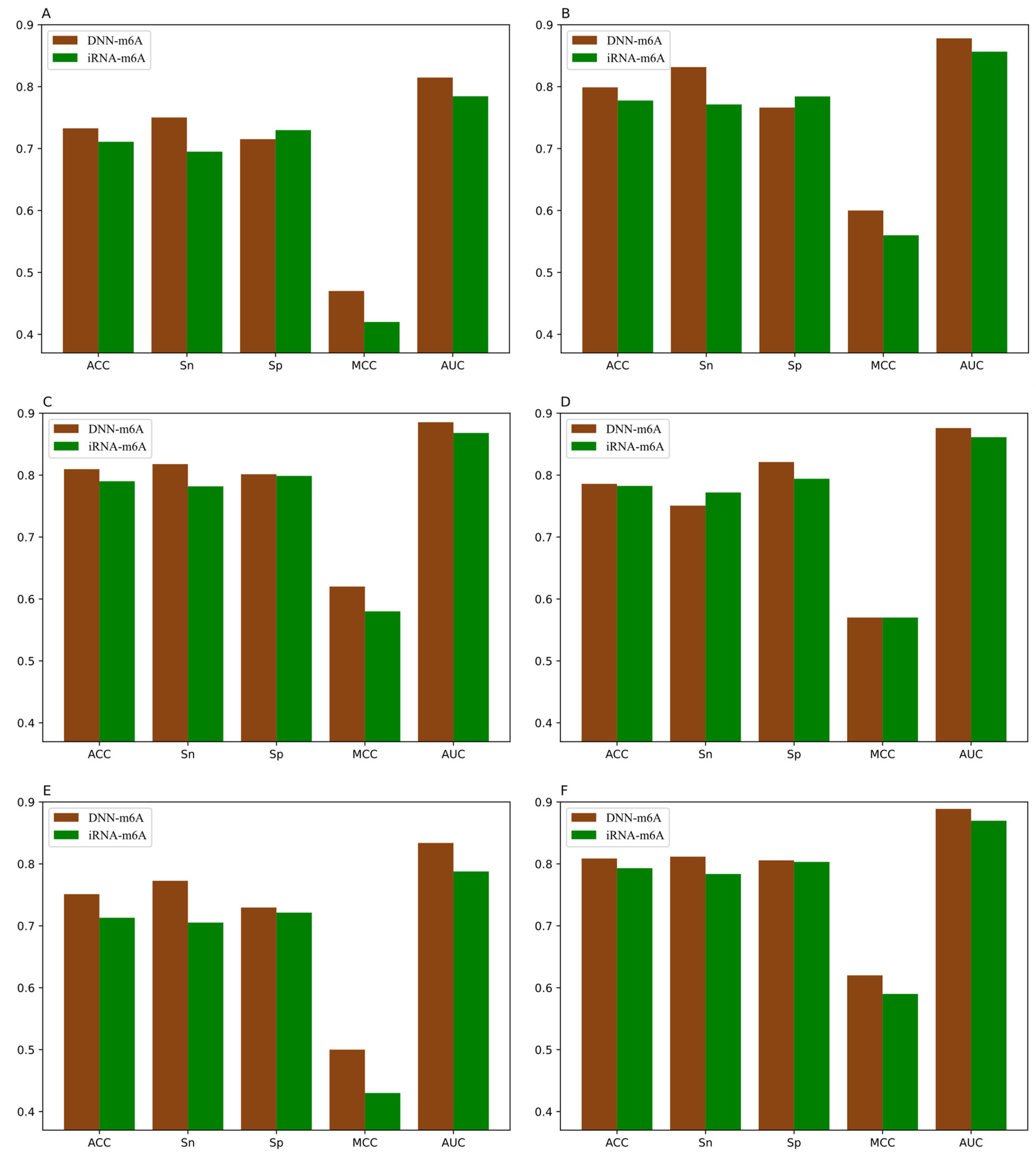
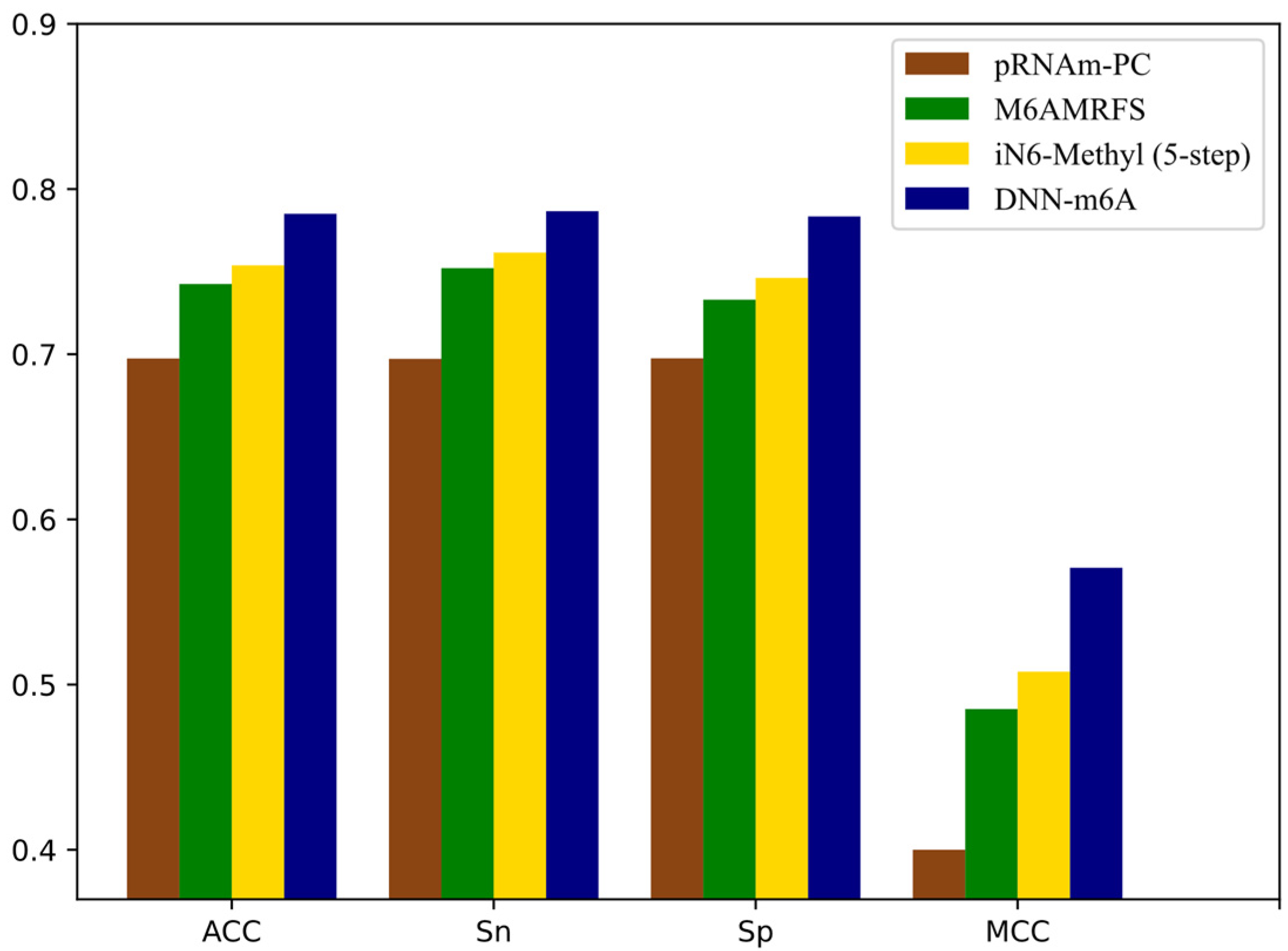
3.5. Comparison of DNN-m6A with Other State-of-the-Art Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Narayan, P.; Rottman, F.M. Methylation of Mrna. In Advances in Enzymology and Related Areas of Molecular Biology; Nord, F.F., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 255–285. [Google Scholar]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 2012, 40, 5023–5033. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Pan, T. RNA epigenetics. Transl. Res. 2015, 165, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Perry, R.P.; Kelley, D.E.; Friderici, K.; Rottman, F. The methylated constituents of L cell messenger RNA: Evidence for an unusual cluster at the 5′ terminus. Cell 1975, 4, 387–394. [Google Scholar] [CrossRef]
- Schibler, U.; Kelley, D.E.; Perry, R.P. Comparison of methylated sequences in messenger RNA and heterogeneous nuclear RNA from mouse L cells. J. Mol. Biol. 1977, 115, 695–714. [Google Scholar] [CrossRef]
- Wei, C.M.; Gershowitz, A. 5’-Terminal and Internal Methylated Nucleotide Sequences in HeLa Cell IRRMA. Biochemistry 1976, 15, 397–407. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; He, C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013, 29, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhao, X.; Wu, Y.-S.; Li, M.-M.; Wang, X.-J.; Yang, Y.-G. N6-methyl-adenosine (m6A) in RNA: An Old Modification with A Novel Epigenetic Function. Genom. Proteom. Bioinform. 2013, 11, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.-G.; et al. N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Bodi, Z.; Button, J.D.; Grierson, D.; Fray, R.G. Yeast targets for mRNA methylation. Nucleic Acids Res. 2010, 38, 5327–5335. [Google Scholar] [CrossRef]
- Zhao, B.S.; Roundtree, I.A.; He, B. Post-transcriptional gene regulation by mRNA modifications. Nat. Rev. Mol. Cell Biol. 2017, 18, 31–42. [Google Scholar] [CrossRef]
- Lin, S.; Choe, J.; Du, P.; Triboulet, R.; Gregory, R.I. The m 6 A Methyltransferase METTL3 Promotes Translation in Human Cancer Cells. Mol. Cell 2016, 62, 335–345. [Google Scholar] [CrossRef]
- Liu, J.; Eckert, M.A.; Harada, B.T.; Liu, S.-M.; Lu, Z.; Yu, K.; Tienda, S.M.; Chryplewicz, A.; Zhu, A.C.; Yang, Y.; et al. m6A mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat. Cell Biol. 2018, 20, 1074–1083. [Google Scholar] [CrossRef]
- Ma, J.; Yang, F.; Zhou, C.; Liu, F.; Yuan, J.; Wang, F.; Wang, T.; Xu, Q.; Zhou, W.; Sun, S. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N 6 -methyladenosine-dependent primary MicroRNA processing. Hepatology 2017, 65, 529–543. [Google Scholar] [CrossRef]
- Chen, X.-Y.; Zhang, J.; Zhu, J.-S. The role of m6A RNA methylation in human cancer. Mol. Cancer 2019, 18, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Keith, G. Mobilities of modified ribonucleotides on two-dimensional cellulose thin-layer chromatography. Biochimie 1995, 77, 142–144. [Google Scholar] [CrossRef]
- Zheng, G.; Dahl, J.A.; Niu, Y.; Fedorcsak, P.; Huang, C.-M.; Li, C.J.; Vågbø, C.B.; Shi, Y.; Wang, W.-L.; Song, S.-H.; et al. ALKBH5 Is a Mammalian RNA Demethylase that Impacts RNA Metabolism and Mouse Fertility. Mol. Cell 2013, 49, 18–29. [Google Scholar] [CrossRef]
- Dominissini, D.; Moshitch-Moshkovitz, S.; Salmon-Divon, M.; Amariglio, N.; Rechavi, G. Transcriptome-wide mapping of N6-methyladenosine by m6A-seq based on immunocapturing and massively parallel sequencing. Nat. Protoc. 2013, 8, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.D.; Saletore, Y.; Zumbo, P.; Elemento, O.; Mason, C.E.; Jaffrey, S.R. Comprehensive Analysis of mRNA Methylation Reveals Enrichment in 3′ UTRs and near Stop Codons. Cell 2012, 149, 1635–1646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zeng, P.; Li, Y.-H.; Zhang, Z.; Cui, Q. SRAMP: Prediction of mammalian N6-methyladenosine (m6A) sites based on sequence-derived features. Nucleic Acids Res. 2016, 44, e91. [Google Scholar] [CrossRef]
- Zhao, Z.; Peng, H.; Lan, C.; Zheng, Y.; Fang, L.; Li, J. Imbalance learning for the prediction of N6-Methylation sites in mRNAs. BMC Genom. 2018, 19, 1–10. [Google Scholar] [CrossRef]
- Chen, W.; Ding, H.; Zhou, X.; Lin, H.; Chou, K.-C. iRNA(m6A)-PseDNC: Identifying N6-methyladenosine sites using pseudo dinucleotide composition. Anal. Biochem. 2018, 561–562, 59–65. [Google Scholar] [CrossRef]
- Chen, W.; Xing, P.; Zou, Q. Detecting N6-methyladenosine sites from RNA transcriptomes using ensemble Support Vector Machines. Sci. Rep. 2017, 7, 40242. [Google Scholar] [CrossRef]
- Xing, P.; Su, R.; Guo, F.; Wei, L. Identifying N6-methyladenosine sites using multi-interval nucleotide pair position specificity and support vector machine. Sci. Rep. 2017, 7, srep46757. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Chen, H.; Su, R. M6APred-EL: A Sequence-Based Predictor for Identifying N6-methyladenosine Sites Using Ensemble Learning. Mol. Ther. Nucleic Acids 2018, 12, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yan, R. RFAthM6A: A new tool for predicting m6A sites in Arabidopsis thaliana. Plant Mol. Biol. 2018, 96, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Akbar, S.; Hayat, M. iMethyl-STTNC: Identification of N6-methyladenosine sites by extending the idea of SAAC into Chou’s PseAAC to formulate RNA sequences. J. Theor. Biol. 2018, 455, 205–211. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, X.; Yu, D.-J.; Jia, J.; Qiu, W.-R.; Chou, K.-C. pRNAm-PC: Predicting N6-methyladenosine sites in RNA sequences via physical–chemical properties. Anal. Biochem. 2016, 497, 60–67. [Google Scholar] [CrossRef]
- Qiang, X.; Chen, H.; Ye, X.; Su, R.; Wei, L. M6AMRFS: Robust Prediction of N6-Methyladenosine Sites with Sequence-Based Features in Multiple Species. Front. Genet. 2018, 9, 495. [Google Scholar] [CrossRef]
- Dao, F.-Y.; Lv, H.; Yang, Y.-H.; Zulfiqar, H.; Gao, H.; Lin, H. Computational identification of N6-methyladenosine sites in multiple tissues of mammals. Comput. Struct. Biotechnol. J. 2020, 18, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, L.-Q.; Zhao, Y.-L.; Yang, C.-G.; Roundtree, I.A.; Zhang, Z.; Ren, J.; Xie, W.; He, C.; Luo, G.-Z. Single-base mapping of m6A by an antibody-independent method. Sci. Adv. 2019, 5, eaax0250. [Google Scholar] [CrossRef]
- Zou, Q.; Lin, G.; Jiang, X.; Liu, X.; Zeng, X. Sequence clustering in bioinformatics: An empirical study. Briefings Bioinform. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.E. (Ed.) Encyclopedia of Metagenomics; Springer: Boston, MA, USA, 2015. [Google Scholar]
- Feng, P.; Ding, H.; Chen, W.; Lin, H. Identifying RNA 5-methylcytosine sites via pseudo nucleotide compositions. Mol. Biosyst. 2016, 12, 3307–3311. [Google Scholar] [CrossRef]
- Li, G.-Q.; Liu, Z.; Shen, H.-B.; Yu, D.-J. TargetM6A: Identifying N6-Methyladenosine Sites from RNA Sequences via Position-Specific Nucleotide Propensities and a Support Vector Machine. IEEE Trans. Nanobiosci. 2016, 15, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Manavalan, B.; Basith, S.; Shin, T.H.; Lee, D.Y.; Wei, L.; Lee, G. 4mCpred-EL: An Ensemble Learning Framework for Identification of DNA N4-methylcytosine Sites in the Mouse Genome. Cells 2019, 8, 1332. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Tian, B.; Liu, Y.; Zhang, Y.; Ma, Q.; Yu, B. StackRAM: A cross-species method for identifying RNA N 6 -methyladenosine sites based on stacked ensembl. Bioinform. Prepr. Apr. 2020. [Google Scholar] [CrossRef]
- Xu, Z.-C.; Wang, P.; Qiu, W.-R.; Xiao, X. iSS-PC: Identifying Splicing Sites via Physical-Chemical Properties Using Deep Sparse Auto-Encoder. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Fang, T.; Zhang, Z.; Sun, R.; Zhu, L.; He, J.; Huang, B.; Xiong, Y.; Zhu, X. RNAm5CPred: Prediction of RNA 5-Methylcytosine Sites Based on Three Different Kinds of Nucleotide Composition. Mol. Ther. Nucleic Acids 2019, 18, 739–747. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Z.; Mao, X.; Li, Q. m7GPredictor: An improved machine learning-based model for predicting internal m7G modifications using sequence properties. Anal. Biochem. 2020, 609, 113905. [Google Scholar] [CrossRef]
- Chen, W.; Feng, P.; Tang, H.; Ding, H.; Lin, H. Identifying 2′-O-methylationation sites by integrating nucleotide chemical properties and nucleotide compositions. Genomics 2016, 107, 255–258. [Google Scholar] [CrossRef]
- Chen, W.; Yang, H.; Feng, P.; Ding, H.; Lin, H. iDNA4mC: Identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics 2017, 33, 3518–3523. [Google Scholar] [CrossRef]
- Feng, P. iRNA-PseColl: Identifying the Occurrence Sites of Different RNA Modifications by Incorporating Collective Effects of Nucleotides into PseKNC. Mol. Ther. 2017, 7, 155–163. [Google Scholar] [CrossRef]
- Xiang, S.; Liu, K.; Yan, Z.; Zhang, Y.; Sun, Z. RNAMethPre: A Web Server for the Prediction and Query of mRNA m6A Sites. PLoS ONE 2016, 11, e0162707. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, Y.; Li, L.; Liu, Z.; Yang, X.; Yu, D.-J. Accurate RNA 5-methylcytosine site prediction based on heuristic physical-chemical properties reduction and classifier ensemble. Anal. Biochem. 2018, 550, 41–48. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, Y.; Ning, Q.; Zhang, H.; Ji, J.; Yin, M. Identifying N6-methyladenosine sites using extreme gradient boosting system optimized by particle swarm optimizer. J. Theor. Biol. 2019, 467, 39–47. [Google Scholar] [CrossRef]
- He, J.; Fang, T.; Zhang, Z.; Huang, B.; Zhu, X.; Xiong, Y. PseUI: Pseudouridine sites identification based on RNA sequence information. BMC Bioinform. 2018, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wen, X.; Shao, X.-J.; Deng, N.-Y.; Chou, K.-C. iHyd-PseAAC: Predicting Hydroxyproline and Hydroxylysine in Proteins by Incorporating Dipeptide Position-Specific Propensity into Pseudo Amino Acid Composition. Int. J. Mol. Sci. 2014, 15, 7594–7610. [Google Scholar] [CrossRef] [PubMed]
- Zou, H.; Hastie, T. Addendum: Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B 2005, 67, 768. [Google Scholar] [CrossRef]
- Saunders, C.; Gammerman, A.; Vovk, V. Ridge Regression Learning Algorithm in Dual Variables. 1998. Available online: https://eprints.soton.ac.uk/258942/1/Dualrr_ICML98.pdf (accessed on 27 February 2021).
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Thornton, C.; Hutter, F.; Hoos, H.H.; Leyton-Brown, K. Auto-WEKA: Combined selection and hyperparameter optimization of classification algorithms. In Proceedings of the 19th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining—KDD ’13, Chicago, IL, USA, 11–14 August 2013; p. 847. [Google Scholar] [CrossRef]
- Xia, Y.; Liu, C.; Li, Y.; Liu, N. A boosted decision tree approach using Bayesian hyper-parameter optimization for credit scoring. Expert Syst. Appl. 2017, 78, 225–241. [Google Scholar] [CrossRef]
- Bergstra, J.S.; Bardenet, R.; Bengio, Y.; Kégl, B. Algorithms for Hyper-Parameter Optimization. Available online: https://core.ac.uk/download/pdf/46766638.pdf (accessed on 27 February 2021).
- Sokolova, M.; Lapalme, G. A systematic analysis of performance measures for classification tasks. Inf. Process. Manag. 2009, 45, 427–437. [Google Scholar] [CrossRef]
- Roweis, S.T. Nonlinear Dimensionality Reduction by Locally Linear Embedding. Science 2000, 290, 2323–2326. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Long, F.; Ding, C. Feature selection based on mutual information criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005, 27, 1226–1238. [Google Scholar] [CrossRef] [PubMed]
- Ng, A.Y.; Jordan, M.I.; Weiss, Y. On Spectral Clustering: Analysis and an algorithm. Adv. Neural Inf. Process. Syst. 2002, 2, 849–856. [Google Scholar]
- Wall, M.E.; Rechtsteiner, A.; Rocha, L.M. Singular Value Decomposition and Principal Component Analysis. In A Practical Approach to Microarray Data Analysis; Springer: Boston, MA, USA, 2003; p. 19. [Google Scholar]
- Nazari, I.; Tahir, M.; Tayara, H.; Chong, K.T. iN6-Methyl (5-step): Identifying RNA N6-methyladenosine sites using deep learning mode via Chou’s 5-step rules and Chou’s general PseKNC. Chemom. Intell. Lab. Syst. 2019, 193, 103811. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Tissues | Positive | Negative | ||
|---|---|---|---|---|---|
| Training | Testing | Training | Testing | ||
| Human | Brain | 4605 | 4604 | 4605 | 4604 |
| Kidney | 4574 | 4573 | 4574 | 4573 | |
| Liver | 2634 | 2634 | 2634 | 2634 | |
| Mouse | Brain | 8025 | 8025 | 8025 | 8025 |
| Heart | 2201 | 2200 | 2201 | 2200 | |
| Kidney | 3953 | 3952 | 3953 | 3952 | |
| Liver | 4133 | 4133 | 4133 | 4133 | |
| Testis | 4707 | 4706 | 4707 | 4706 | |
| Rat | Brain | 2352 | 2351 | 2352 | 2351 |
| Kidney | 3433 | 3432 | 3433 | 3432 | |
| Liver | 1762 | 1762 | 1762 | 1762 | |
| Chemical Property | Class | Nucleotides |
|---|---|---|
| Ring Structure | Purine | A, G |
| Pyrimidine | C, U | |
| Functional Group | Amino | A, C |
| Keto | G, U | |
| Hydrogen Bond | Strong | C, G |
| Weak | A, U |
| Hyper-Parameters | Meaning | Search Ranges |
|---|---|---|
| layers | number of hidden layers | (2,3) |
| hidden_1 | number of neurons in the first hidden layer | (100, 800) |
| hidden_2 | number of neurons in the second hidden layer | (50, 700) |
| hidden_3 | number of neurons in the third hidden layer | (25, 600) |
| activation | activation function | elu, selu; softplus; softsign; relu; tanh; hard_sigmoid |
| optimizer | Per-parameter adaptive | RMSprop; Adam; Adamax; SGD; Nadam; Adadelta; Adagrad |
| learning_rate | learning rate of the optimizer | (0.001, 0.09) |
| kernel_initializer | layers weight initializer | uniform; normal; lecun_uniform; glorot_uniform; glorot_normal; he_normal; he_uniform |
| dropout | dropout rate | (0.1, 0.6) |
| epochs | number of iterations | 10; 20; 30; 40; 50; 60; 70; 80; 90; 100 |
| batch_size | number of samples for one training | 40; 50; 60; 70; 80 |
| Hyper- Parameters | H_B | H_K | H_L | M_B | M_H | M_K | M_L | M_T | R_B | R_K | R_L |
|---|---|---|---|---|---|---|---|---|---|---|---|
| layers | 2 | 2 | 3 | 3 | 3 | 2 | 3 | 2 | 2 | 2 | 2 |
| hidden_1 | 116 | 381 | 798 | 576 | 506 | 400 | 794 | 431 | 203 | 316 | 627 |
| hidden_2 | 697 | 147 | 694 | 132 | 621 | 498 | 506 | 217 | 116 | 177 | 234 |
| hidden_3 | - | - | 464 | 598 | 501 | - | 329 | - | - | - | - |
| activation | softplus | selu | softsign | softplus | softsign | selu | selu | softplus | softplus | softplus | hard_sigmoid |
| optimizer | Adagrad | Adamax | Adadelta | Adagrad | Adadelta | Adagrad | Adagrad | SGD | Adamax | Adadelta | Adadelta |
| learning_rate | 0.0373 | 0.0042 | 0.0441 | 0.0479 | 0.0587 | 0.0015 | 0.0026 | 0.0860 | 0.0027 | 0.0667 | 0.0899 |
| kernel_initializer | glorot_normal | glorot_normal | lecun_uniform | lecun_uniform | uniform | uniform | lecun_uniform | glorot_uniform | he_uniform | he_normal | he_uniform |
| dropout | 0.3233 | 0.4596 | 0.5073 | 0.2525 | 0.5971 | 0.4981 | 0.2401 | 0.4129 | 0.3226 | 0.1214 | 0.1840 |
| epochs | 70 | 10 | 100 | 20 | 70 | 30 | 50 | 80 | 20 | 50 | 30 |
| batch_size | 80 | 80 | 50 | 70 | 70 | 60 | 70 | 80 | 80 | 80 | 50 |
| Species | Tissues | TPE | ACC | Sn | Sp | MCC | AUC |
|---|---|---|---|---|---|---|---|
| Human | Brain | Yes | 0.7378 | 0.7848 | 0.6908 | 0.4788 | 0.8165 |
| No | 0.7344 | 0.8165 | 0.6523 | 0.4764 | 0.8131 | ||
| Kidney | Yes | 0.8048 | 0.8356 | 0.7739 | 0.6107 | 0.8841 | |
| No | 0.7984 | 0.8640 | 0.7328 | 0.6023 | 0.8826 | ||
| Liver | Yes | 0.8130 | 0.8219 | 0.8041 | 0.6264 | 0.8905 | |
| No | 0.8077 | 0.8466 | 0.7688 | 0.6188 | 0.8859 | ||
| Mouse | Brain | Yes | 0.7936 | 0.8176 | 0.7697 | 0.5880 | 0.8778 |
| No | 0.7890 | 0.8160 | 0.7621 | 0.5807 | 0.8758 | ||
| Heart | Yes | 0.7617 | 0.7751 | 0.7483 | 0.5238 | 0.8439 | |
| No | 0.7565 | 0.7865 | 0.7265 | 0.5144 | 0.8375 | ||
| Kidney | Yes | 0.8196 | 0.8320 | 0.8072 | 0.6396 | 0.8953 | |
| No | 0.8151 | 0.8530 | 0.7771 | 0.6331 | 0.8944 | ||
| Liver | Yes | 0.7358 | 0.7757 | 0.6959 | 0.4733 | 0.8139 | |
| No | 0.7303 | 0.8210 | 0.6397 | 0.4697 | 0.8114 | ||
| Testis | Yes | 0.7662 | 0.8099 | 0.7225 | 0.5347 | 0.8493 | |
| No | 0.7616 | 0.8007 | 0.7225 | 0.5259 | 0.8429 | ||
| Rat | Brain | Yes | 0.7827 | 0.7908 | 0.7746 | 0.5658 | 0.8678 |
| No | 0.7819 | 0.8180 | 0.7457 | 0.5657 | 0.8672 | ||
| Kidney | Yes | 0.8338 | 0.8427 | 0.8249 | 0.6679 | 0.9104 | |
| No | 0.8321 | 0.8488 | 0.8153 | 0.6658 | 0.9087 | ||
| Liver | Yes | 0.8263 | 0.8417 | 0.8110 | 0.6533 | 0.8991 | |
| No | 0.8229 | 0.8428 | 0.8031 | 0.6474 | 0.8962 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, L.; Qin, X.; Liu, M.; Xu, Z.; Liu, G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes 2021, 12, 354. https://doi.org/10.3390/genes12030354
Zhang L, Qin X, Liu M, Xu Z, Liu G. DNN-m6A: A Cross-Species Method for Identifying RNA N6-methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes. 2021; 12(3):354. https://doi.org/10.3390/genes12030354
Chicago/Turabian StyleZhang, Lu, Xinyi Qin, Min Liu, Ziwei Xu, and Guangzhong Liu. 2021. "DNN-m6A: A Cross-Species Method for Identifying RNA N6-methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion" Genes 12, no. 3: 354. https://doi.org/10.3390/genes12030354
APA StyleZhang, L., Qin, X., Liu, M., Xu, Z., & Liu, G. (2021). DNN-m6A: A Cross-Species Method for Identifying RNA N6-methyladenosine Sites Based on Deep Neural Network with Multi-Information Fusion. Genes, 12(3), 354. https://doi.org/10.3390/genes12030354