Abstract
Glioblastoma (GBM) is the most common primary malignant brain tumour in adults. Despite the combination of novel therapeutical approaches, it remains a deadly malignancy with an abysmal prognosis. GBM is a polymorphic tumour from both molecular and histological points of view. It consists of different malignant cells and various stromal cells, contributing to tumour initiation, progression, and treatment response. GBM’s microenvironment is multifaceted and is made up of soluble factors, extracellular matrix components, tissue-resident cell types (e.g., neurons, astrocytes, endothelial cells, pericytes, and fibroblasts) together with resident (e.g., microglia) or recruited (e.g., bone marrow-derived macrophages) immune cells. These latter constitute the so-called immune microenvironment, accounting for a substantial GBM’s tumour volume. Despite the abundance of immune cells, an intense state of tumour immunosuppression is promoted and developed; this represents the significant challenge for cancer cells’ immune-mediated destruction. Though literature data suggest that distinct GBM’s subtypes harbour differences in their microenvironment, its role in treatment response remains obscure. However, an in-depth investigation of GBM’s microenvironment may lead to novel therapeutic opportunities to improve patients’ outcomes. This review will elucidate the GBM’s microenvironment composition, highlighting the current state of the art in immunotherapy approaches. We will focus on novel strategies of active and passive immunotherapies, including vaccination, gene therapy, checkpoint blockade, and adoptive T-cell therapies.
1. Introduction
Brain tumours are characterized by high mortality due to their localization and invasive growth [1]. Glioblastoma (GBM) accounts for about 70% of gliomas and 15% of primary brain tumours, representing the most frequent primary malignant brain tumour in adults [2]. Recent attention to stem-like glioma cells has identified neural stem cells (NSCs), a subfraction of cells limited to germinal areas, as precursor cells in gliomas [3]. GBM consists of a heterogeneous population made of malignant cells and different stromal cells, which altogether contribute to tumour formation or progression and response to treatment [4]. The current standard of care of GBM contemplates surgical resection, radiotherapy, and chemotherapy with temozolomide (TMZ) [5]. Over the past few decades, improvements in outcome have been modest; however, new approaches (including gene therapy or targeted therapies—such as monoclonal antibodies or tyrosine kinase inhibitors together with immunotherapeutic strategies) may display more significant potential. In addition to efforts that have led to an improved understanding of the genetic basis of gliomas, a considerable body of work has indicated an increasing recognition of the interaction between glioma and immunity [6]. The significant survival advantage of immunotherapy in numerous extracranial tumours encouraged experimental studies in glioblastoma patients [7]. Data suggest that glioma cells develop mechanisms to evade the immune system [6]; in turn, the tumour microenvironment promotes a state of immunosuppression, offering new therapeutic chances that may improve the outcome of patients [4]. This review aims to elucidate the composition of GBM’s microenvironment and to highlight the current state of the art of novel immunotherapeutic approaches, with a particular focus on active and passive immunotherapies strategies, including vaccination, gene therapy, checkpoint blockade and adoptive T-cell therapies.
2. Epidemiology
Approximately 80% of all brain malignant tumours are gliomas, of which around 70% are GBM [8].
GBM occurs mainly in men with a median age of 64 years (1.3–1.6:1 compared to women), particularly in whites of European descent (2:1 compared to African Americans). The annual incidence ranges between 3 and 5 newly diagnosed cases per 100,000 inhabitants [9].
Notwithstanding the variety of current therapies, GBM remains a deadly disease with a tremendously poor prognosis: patients survive approximately 15 months after diagnosis, and only 5.5% of these are alive five years after diagnosis [10]. Clinical and biological parameters such as tumour size and location, age at presentation, Karnofsky performance score (KPS) at presentation, histologic findings and molecular genetic factors or therapeutic approaches undertaken affect survival in GBM patients [11].
3. Histological and Molecular Classification
According to morphological similarities with neuroglial cells in the normal brain, gliomas have been traditionally classified into astrocytomas, oligodendrogliomas, and ependymomas [9]. The World Health Organization (WHO) classifies astrocytomas based on histologic features into four grades of prognosis, of which anaplastic astrocytoma (grade III) and GBM (grade IV) are considered malignant gliomas [12]. GBM exhibits a pleomorphic cell population of small, poorly differentiated and large multinucleated tumour cells with brisk mitotic activity in the context of multifocal necrosis (with pseudopalisading nuclei) and glomeruloid microvascular proliferation [10]. Although GBM is one of the most heterogeneous neoplasms in humans, only three morphologic rare variants are recognized in the 2016 WHO Classification of Tumors of the Central Nervous System (CNS): gliosarcoma, giant cell glioblastoma, and epithelioid glioblastoma [13]. Malignant gliomas typically contain both neoplastic cells and stroma, contributing to the tumour’s histologic heterogeneity and variable outcome. Based on gene expression profiles, GBM can be further classified into four subtypes, such as classical, proneural, neural, and mesenchymal, conferring differences in terms of aggressiveness, potential to progress, and thus diverse prognosis to the tumour [14]. Histopathological classification is currently assisted by molecular genetic studies that provide diagnostic, prognostic, and predictive values [14]. In detail, three markers have undergone extensive studies in recent years: (i) 1p/19q chromosomal codeletion; (ii) O6-methylguanine methyltransferase (MGMT) promoter methylation; and (iii) mutations of isocitrate dehydrogenase (IDH) 1 and 2 [15]. According to the expression of these markers, gliomas of adulthood are classified into three main groups: (i) IDH-mutant, 1p19q co-deleted tumours, with a predominantly oligodendroglial morphology and favourable outcome; (ii) IDH-mutant, non-1p19q co-deleted tumours, with an astrocytic morphology and intermediate outcome; and (iii) IDH wild-type tumours, which are mostly glioblastomas, with an unfavourable prognosis [16]. In the 2016 WHO Classification of Tumors of the CNS, GBMs have been classified as: (i) glioblastoma IDH-wildtype (about 90% of cases), which corresponds to the clinically defined primary or de novo glioblastoma and occurs in patients over 55 years of age; (ii) glioblastoma IDH-mutant (about 10% of cases), which corresponds to the clinically defined secondary glioblastoma (characterized by a history of prior lower grade diffuse gliomas) and preferentially arises in younger patients; and (iii) glioblastoma NOS, for which full IDH evaluation cannot be performed [13,17]. Primary and secondary GBMs constitute distinct diseases that develop through different genetic pathways and differ in prognosis and therapy response. Primary glioblastomas typically display EGFR overexpression, CDKN2A (p16) deletions, PTEN (MMAC1) mutations, and MDM2 amplification. In contrast, secondary glioblastomas often have TP53 mutations as the earliest alteration [18].
4. Barriers of the CNS
The brain was long believed to be widely disregarded by the immune system because of two morphological peculiarities: the absence of lymphatic vessels and the presence of the blood-brain barrier (BBB) [19]. Two physical barriers regulate the moving of immune cells from the blood to the CNS: the BBB formed by the CNS parenchymal microvessels and the blood-cerebrospinal fluid (CSF) barrier formed by the choroid plexuses [20]. The BBB is a selectively permeable barrier composed of endothelial cells, pericytes, and astrocytes, which shields the brain from pathogens, toxins and inflammatory cells within the peripheral circulation [19]. In physiologic conditions, the brain hosts an immunosuppressive milieu that protects the delicate (and non-regenerative) neural tissue from inflammatory insults [4]. Leukocytes entry into the CNS is restricted due to the tight junctions of the BBB at the capillary segment of the vascular arbour. Leukocytes recruitment occurs in postcapillary venules, and three distinct entry routes may be defined: (i) from blood to CSF, across the choroid plexus; (ii) from blood to the subarachnoid space, through meningeal vessels; and (iii) from blood to parenchymal perivascular spaces [21]. In experimental autoimmune encephalomyelitis, it has been demonstrated that mononuclear cells’ diapedesis occurs via the transendothelial process of the cerebral venules, keeping the endothelial tight junctions intact [22]. Also, CSF circulation has been revised by the discoveries over the glymphatic system, involving paravascular routes where fluids are freely exchanged between the brain’s cerebral interstitial fluid (ISF) and CSF, without crossing the endothelial cell layer [23,24,25]. Meningeal lymphatic vessels receive CSF from the adjacent subarachnoid space and ISF via the glymphatic system, then transporting the fluids into deep cervical lymph nodes via foramina at the skull base [26]. Thus, the BBB is not designed to keep leukocytes out of the brain but contributes to regulating immunoregulatory cells and molecules in the CNS microenvironment [27]. Also, the CNS immune privilege may be explained by the lack of dendritic cells (DCs) or other resident antigen-presenting cells (APCs) capable of conveying antigens to lymph nodes outside the CNS parenchyma. This characteristic makes the CNS virtually resilient to constructing immune reactions [28].
5. Tumor Microenvironment in Glioblastoma
One of the main explanations for treatment resistance and tumour recurrence in GBM resides in the big changes within the tumour microenvironment (TME) [29]. TME is made up of numerous cell types: (i) tissue-resident cells such as neurons and astrocytes; (ii) myeloid cells such as resident microglia; (iii) bone marrow-derived macrophages (BMDMs), bone marrow-derived DCs and neutrophils; (iv) other immune cells as lymphoid cells; (v) endothelial cells, pericytes, and fibroblasts. All these cells are surrounded by a distinctive extra-cellular matrix (ECM) [30,31,32,33]. Bidirectional interaction between cells and their microenvironment is crucial for both normal tissue homeostasis and tumour growth. In particular, tumour cells interact with the associated stroma and influence disease initiation, progression, and patient outcome [34]. A state of immunosuppression characterizes GBM’s TME, thanks to the secretion of several cytokines by tumour cells, microglia, and tumour-associated macrophages (TAMs) [32]. GBM cells produce cytokines, chemokines, growth factors and ECM modifying enzymes, extracellular vesicles and proteins to construct a favourable tumour microenvironment [29]. Also, cells in TME interact with each other and with the neoplastic cells through different suppressor receptors like PD-1, CTLA-4, CD70 and gangliosides that increase the tumour immune escaping [35,36]. This results in: (i) the inhibition of the immune response; (ii) the activation of FoxP3+ regulatory T-cells (Tregs); (iii) the suppression of NK activity and T-cell activation; (iv) the induction of T-cell apoptosis; (v) the switch to an M2 immunosuppressive phenotype of TAMs; and (vi) the downregulation of MHC expression [37,38].
A crucial point of the brain’s immune privilege is linked to the resident cells’ intrinsic capacity to induce apoptosis of infiltrating T-cells. This process is driven especially by adult astrocytes representing the first line of defence of the BBB against the entry of activated T-cells in CNS [39,40]. Indeed, astrocytes express death-ligand CD95L (APO-1L/FasL) to kill CD95+ infiltrating T-cells [41,42]. In gliomas, the BBB is compromised, enabling the infiltration of immune cells [29,43].
5.1. Cells in GBM TME
5.1.1. Tumour-Infiltrating Lymphocytes-TILs
The majority of the Tumor-infiltrating lymphocytes (TILs) are represented by CD8+ CTLs, CD4+ T helper cells, and by regulatory CD4+/FoxP3+ T-cells (Tregs) [44,45].
CD3+ and CD8+ T-cell infiltration in GBM’s TME increases from low-grade to high-grade tumors, maybe due to the disruption of the BBB, and at the same time it has been correlated to better patients’ prognosis, independently from age, MGMT promoter methylation, and post-operative treatment [44,46].
In general, GBM induces a state of global T-cell impairment and functional immunosuppression, exploiting several different mechanisms such as senescence, tolerance, anergy, exhaustion, and ignorance [45]. Indeed, GBM patients display a reduction of systemic CD4+ T-cells levels together with an upregulation of inhibiting receptors, such as CTLA-4, CD73, and CD39, leading to a decrease in T-cells activity [44].
5.1.2. Regulatory T Cells-Tregs
Immune tolerance is mediated mainly by regulatory T-cells (Tregs), immunosuppressive T-cells, which negatively module the immune response and protect against autoimmunity [47,48,49].
Two types of Tregs with different origins are present in CNS: the thymus-derived natural Tregs (nTregs) that account for most of the brain-tumour resident Tregs and the induced Treg (iTregs) [50,51,52]. In particular, GBM tumour cells recruit nTregs [51,53]. Tregs drives tumour immune microenvironment, tumour progression and patient outcome [50,51,52]. Indeed, GBM immune-escape ability is associated with the presence of Tregs, and their number is related to glioma’s grade. However, exploited mechanisms have been only partially understood [48,49,54]. Several studies have demonstrated how a large amount of CD4+ cells is represented by Tregs, which downregulate the IFNγ-based antitumour response thanks to the production of Th-2 polarizing cytokines, such as IL-10 and TGFβ [47,55,56,57,58]. TGF-βs (β1 and β2) may hamper T-cell recruitment in CNS through the downregulation of ICAM-1 and VCAM-1; this finding opens the possibility to use anti-TGF-βs drugs to enhance immunotherapy efficacy [46].
Tregs recruitment occurs through the binding of CCR4 on Tregs surface and CCL22 secreted by GBM tumour cells and CD163+ TAMs, under the induction of tumour-derived CCL20 and osteoprotegerin [59,60]. Another mechanism involved in Tregs chemotaxis is the expression of the tumour-enzyme indoleamine 2,3 dioxygenase (IDO), which has been associated with tumour progression and lower survival-rates [61,62].
Several target therapies against Tregs have been tested in combination with standard chemotherapy, such as mAb against IL-2Rα (CD25) and CTLA-4, cytokines inhibitors or GITR agonists, up-regulated in Treg and activated T-cells. However, only low levels of response have been reported with the administration of these innovative drugs [47,57,58,63,64,65,66,67].
5.1.3. CAR-T
Recently, the broad term “T cell exhaustion” has been used to describe not only the response of T cells to chronic antigen stimulation in the setting of chronic viral infection but also in response to tumours. T-cell exhaustion is tightly linked to the TME and the production of several concomitant key-factors: (i) the upregulation of inhibiting receptors (PD-1, Tim-3, CD39, TIGIT, Lag-3 and CTLA-4); (ii) the presence of cytokines’ dysfunction, with high levels of immunosuppressive soluble mediators (IL-10, TGF-B, PGE2 and CSF-1); and (iii) the recruitment of immunosuppressive cell species, such as MDSC or Tregs [68,69].
A therapy based on genetically engineered T-cells with Chimeric Antigen Receptor (CAR) has been developed to increase the tumour immunogenicity. CARs are artificial fusion proteins engineered to bind to specific tumour antigens via a single-chain fragment variable (scFv) [70,71] This allows inducing an anti-tumour T-cell response, overcoming the low tumour mutational load, the defective neoantigens presentation, and the lack of immune priming in GBM [72,73,74].
Significant results were obtained by anti-CD19 CAR-T cells in the context of hematologic tumours [75,76,77]. Pre-clinical and early phase clinical trials have been conducted on several brain tumour targets, such as IL13Rα2, EGFRvIII, HER2, EphA2, GD2, B7-H3, chlorotoxin, and CD70. In particular, three early phase trials targeting IL13Rα2, Her2/CMV, and EGFRvIII have been performed, reporting good safety and tolerability rates, with a promising anti-tumour response [78,79,80,81]. Furthermore, HER2, EGFRvIII, and IL-13 receptor α 2 (IL-13 Rα2) are also exposed on the surface of cancer stem cells (CSCs), paving the way for new possible application of CAR-T therapy in cancer immunology [82].
A more profound comprehension of cellular heterogeneity and the possibility of combining other immunotherapeutic approaches may overcome CAR-T tumour resistance.
5.1.4. Microglia and Tumour-Associated Macrophages-TAMs
During embryonic life, some progenitor cells from the yolk sac migrate into the CNS and differentiate into parenchymal microglia, maintained by self-renewal during adult life [83,84,85,86].
Microglia preserves neuroimmune homeostasis, supporting the axon’s development and contributing to the elimination of apoptotic cells, but also plays a pivot role in cancer setting [87].
In GBM, tumour-associated macrophages (TAMs) may have two different origins: (i) tumour-infiltrating monocytes, which derive from the hematopoietic stem cells (bone marrow-derived macrophages-BMDMs); and (ii) tissue-resident macrophages. Among this latter group, two further subpopulations can be recognized: (i) the above mentioned parenchymal microglia; and (ii) the peripheral brain macrophages located in the meninges, choroid plexuses and the perivascular sites [35,83,84,85,86,87,88,89].
According to the TAMs’ origin, GBM’s TME reveals different expression levels of proteins involved in antigen presentation and cytokines production, such as IL-1 [90,91].
This could explain the ability of GBM’s TME to select functionally different TAMs populations that may play a different role in the context of the tumour growth, as demonstrated in other malignancies, such as lung and breast cancer [92,93].
In GBM tissue, TAMs account for about one-third of TME’s cells. They have been shown to represent a potentially powerful target for targeted therapy [84,94]. Indeed, the number of TAMs is related directly to tumour grade, and inversely to patients’ survival [14,34,95].
To understand the functional role of the different TAMs populations, one of the most significant challenges to face is represented by the segregation between microglia and BMDMs, through specific biomarkers [96,97,98].
These two different cell populations share positivity for CD68, CXCR1, F4/80, and IBA1 [99]. Though, transcriptional profiling and in vivo 2-photon microscopy clearly demonstrated the profound molecular differences between these two populations [83,84,100]. In the CNS, microglia commonly does not express CD45; however, it expresses CD45 in inflammatory conditions, becoming hardly distinguishable from BMDMs [35,101]. Likewise, CXCR3 is expressed only by microglia; however, it is also expressed by BMDMs within brain tumours [83].
Conversely, microglia could be discriminated by the positivity for TMEM19, P2RY12, Sall1, and by the low expression of CD206 [102,103,104]. Instead, BMDMs expose on their surface CD44, CD169, and high expression of CD45 and CD206 [84,105].
The binding of microglia to immunogenic antigens like lipopolysaccharide through specific surface receptors, such as TLRs, NODs, SR, leads to its activation towards a pro-inflammatory M1 phenotype, characterized by: (i) the activation of enzymes, such as NADPH oxidase and inducible nitric oxide synthase (iNOS); (ii) the increased transcription of STAT1, NF-Kβ, TNF-α; (iii) the secretion of pro-inflammatory chemokines and cytokines; (iv) the acquisition of antigen presentation capacity, thanks to the expression of MHCI and costimulatory molecules like CD40, CD80, and CD86 [30,106,107,108].
The transition to an M2-like anti-inflammatory phenotype is mediated by IL4 and IL13 produced by Th2 lymphocytes, leading to the activation of STAT6, TGF-β and IL10, together with CCL15, -17, -22, and -24 [30].
M2 polarized macrophages are further sub-classified into M2a, M2b, and M2c phenotypes, and express both general (CD68, CD11b, and IBA1) and M2-related markers (CD163, CD204 and CD206) [109,110].
There is a bidirectional interaction between tumour cells and M2c cells. GBM cells produce IL10, TGF-β and glucocorticoids, stimulating the expansion of the M2c population, which in turn promotes the proliferation of tumour cells. Hence, GBM’s TAMs acquire mainly the M2 polarization, and an unbalance interchange of M1-M2 can be detected [109,111,112,113].
These assumptions make immunosuppressive pro-tumoral M2 macrophages a potential therapeutic target, as demonstrated by oncolytic virotherapy, able to revert M2 to M1 phenotype [99].
This innovative approach is based on the injection of engineered viruses into the systemic circulation or directly at the tumour site, inducing the lysis of the tumoral cells [114]. This results in spreading tumoral neoantigens and damage factors that produce a concomitant innate and adaptive immune activation directed against the tumour [114,115,116].
Indeed, some pioneering clinical trials showed how oncolytic viruses could revert to “warm” the immunologically “cold” GBM’s microenvironment, and increase the response of the tumour to checkpoint inhibitors, as well as to the standard treatments [66,117,118].
5.1.5. Dendritic Cells—DCs
Dendritic cells (DCs) are ubiquitous specialized antigen-presenting cells (APCs) that coordinate several immune responses, from self-tolerance to fighting infections [119].
In a cancer setting, mature DCs can show two different behaviours: immunostimulatory or immunosuppressive. Indeed, DCs can drive the immune response against tumour cells. In particular, DCs promote both the cell-mediated anti-tumour T-cells activation and humoral response [120,121,122]. In turn, tumour cells try to shield themselves, producing immunosuppressive molecules able to change the TME composition, hindering DCs activation and migration, and finally evading immune control [121,123]. For example, cancer cells secrete Fibrinogen-like Protein 2 (FGL2) that antagonizes GM-CSF and blocks the cDC1 activation, escaping from immune-control [124,125].
Ovarian cancer represents a peculiar model to study DCs behaviour; in this tumour, DCs show an immunostimulatory phenotype in the early stages, that becomes immunosuppressive in the later stages. In particular, cancer progression is promoted by the lack of an anti-tumour T-cells response lead by DCs [6,126]. On the other hand, tumour-associated DCs (TADCs) can help tumour expansion, promoting new vessel formation and genomic instability [127,128].
According to their different immunophenotypic profile, distinct DCs populations have been found in the TME of different solid tumours, such as conventional DC1 (cDC1) and DC2 (cDC2), plasmacytoid DCs (pDCs), and monocyte-derived DCs (Mo-DCs) [127,129,130].
Each DCs subgroup develops under the control of a specific repertoire of transcription factors, such as IRF8, IRF4, and E2-2. In particular, cDC1 are IRF8-dependent, express CD141 and CCR7, and migrate from the tumour site to draining lymph nodes stimulating the activation of CD8+ T anti-tumoral cells. The presence of these cells in the TME is related to immune-mediated rejection and may represent a specific target to exploit for DC-based tumour therapies [127,131,132]. Indeed, in melanoma patients, cDC1 are recruited by distinct chemokines like Flt3L, CCL5, and XCL1 produced by NK-cells, and are associated with favourable response to anti-PD1 immunotherapy and better outcome [133,134]. Instead, cDC2 are IRF4-dependent, express CD11b, and lead the activation of Th17 cells, as in the vaccination of mice bearing Lewis lung carcinoma, where significant tumour growth decrease has been registered [127,131,135]. A further subtype of DCs is represented by pDCs, which are E2-2 dependent, express CD123 and are activated by viral infections, producing high quantities of interferon and cytokines. Finally, Mo-DCs are CCR2-dependent and originate from bone marrow-derived Ly6Chi monocytes; Mo-DCs perform antigen processing and presentation functions, even with an inefficient activation of T-cells [136,137].
The organization of the DCs in CNS has not been fully elucidated, and many issues on their function in the context of GBM’s TME remain still unsolved.
Flores et al. demonstrated that HSC CCR2+ migrate from bone-marrow to intracranial PD-L1 resistant brain tumours (GBM and medulloblastoma), giving rise to APCs like Mo-DCs, which stimulate the CD8+ T-response against tumour cells. These findings may pave the way for the DC-therapy as an alternative strategy to treat checkpoint blockade resistant tumours [138].
In support of this, some mouse models studies showed that the use of TLR3 and Flt3L enhances DCs’ activation and, in combination with checkpoint blockade therapy, anti-tumour response is improved [139,140].
Wang et al. have demonstrated how DCs exposed to GBM antigens improve the expression of Nrf2, known to correlate to an immunosuppressive state (with an increase in IL-10 and a reduction in CD80, CD86, and IL12). Hence, the inhibition of Nrf2 could intensify the activity of DCs and T-cells against tumour cells [141].
Based on the above findings, the goal of DCs-based therapies would be to promote cDC1 in amplifying tumour-recognition and eradicating tumour cells by the immune system.
This therapeutic strategy is based on loading DCs with GBM-key antigens, thus activating a specific immune response against them [142]. Peptides or tumour lysates together with immune-modulators are used to create vaccines that promote a durable immune activation against GBM [67,142]. Several pre-clinical and clinical studies demonstrated an increase in immunological memory and overall survival, with a good tolerability profile [143,144,145]. However, further clinical trials are needed to evaluate better the efficacy of DC-based therapies in GBM’s treatment.
5.2. Molecules in GBM’s TME
In GBM’s microenvironment, different mediators are secreted by several cell subtypes, which constitute a complex network between immune and neoplastic compartments, such as: (i) cytokines (IL-6, IL-10, VEGF, TGFβ, M-CSF, GM-CSF, bFGF); (ii) prostaglandins (PGE2); (iii) chemokines (CCL20 and CXCL8); (iv) reactive oxygen species; and (v) cellular components like gangliosides [146,147,148].
TME is supported and modelled by tumour cells through growth factors, ECM modifying enzymes, and extracellular vesicles carrying RNA and proteins [29].
In particular, GBM cells secrete many factors like hepatocyte growth factor (HGF), monocyte chemotactic proteins (MCPs), glial neurotrophic derived factors (GNDF), macrophage colony-stimulating factor (M-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), and the transforming growth factor-β (TGF-β) [14,149].
Furthermore, GBM cells recruit and polarize BMDMs in an M2 immunosuppressive phenotype thanks to the secretion of soluble factors (such as osteopontin, postin, and kynurenine), and contribute to the impairment of T-cell function and the establishment of an immunosuppressive state [150,151,152,153].
In turn, tumour-associated macrophages (TAMs) and microglia represent a rich source of chemokines, ECM constituents, angiogenic molecules and growth factors capable of increasing and supporting GBM cells proliferation and infiltration [30].
Novel therapeutic opportunities may derive from discovering the essential pathways and molecules involved in the crosstalk between GBM cells and their microenvironment.
5.2.1. Transforming Growth Factor-β-TGF-β
GBM cells produce the glioblastoma-derived T-cell suppressor factor (G-TsF), a member of the TGF-β family re-named as TGF-β2, which induces immunosuppression by: (i) blocking T-cells activation and proliferation; (ii) downregulating MHC class II expression on CD4+ T-cells; (iii) inhibiting IL-2 production; (iv) suppressing NK-cells activity; and (v) promoting the activation of FoxP3+ regulatory T-cells (Tregs) [33,37,38]. Accordingly, in murine models infected with lymphocytic choriomeningitis virus (LCMV), the injection of recombinant G-TsF/TGF-β2 inhibits the generation of virus-specific cytotoxic T-cells [33,38,154].
TGF-β promotes tumour growth and invasion by supporting and stimulating GBM stem cells’ migration and enhancing angiogenesis; in particular, it inhibits tumorigenesis in early stages, and promotes tumour growth in later stages [33,38,154].
All these mechanisms contribute to the impairment of tumour immune surveillance and open the possibility of immunotherapeutic strategies based on TGF-β signalling pathway inhibition. [155,156].
5.2.2. Prostaglandin E2-PGE2
Prostaglandin E2 (PGE2) is a prostaglandin produced by COX2 and plays an essential role in the angiogenesis of glial tumours, particularly in GBM, which is a highly vascularized tumour with angiogenesis-dependent growth [157]. Indeed, the increased expression of COX-2 has been correlated with gliomas’ grade [158].
In GBM’s microenvironment, PGE2 concentration is not high enough to suppress T-cell functions on its own. However, PGE2 works together with TGF-β, transforming DCs into regulatory cells and suppressing T-cell proliferation [38].
New antiangiogenetic therapies are based on the oral administration of specific Cox-2 inhibitors that reduce angiogenesis and tumour growth combined with low-dose scheduling of chemotherapeutic drugs [159].
5.2.3. Interleukin-10-IL-10
IL-10 is secreted by immune cells as well as by GBM cells and enhances tumour growth, inhibits the production of interferon-γ (IFN-γ) and tumour necrosis factor-α (TNF-α) by the immune system, and suppresses the antigen presentation process [33,160,161].
In vitro, it has been demonstrated that IL-10 upregulates KPNA2 (a gene coding for nuclear import of proteins); at the same time, knockdown experiments showed that glioma cell growth and invasion were significantly reduced, suggesting IL-10 as a potential target for glioma patients’ treatment [162].
5.2.4. Colony Stimulating Factor 1-CSF-1
Colony-stimulating factor 1 (CSF-1) is a crucial chemokine for TAMs differentiation and survival. CSF-1 works in combination with EGFR, promoting GBM cells invasion [163].
The inhibition of CSF-1 and EGFR prevents and reduces tumour invasion, resulting in a significant improvement in patients’ survival and clinical outcome [94,164].
5.2.5. Cluster of Differentiation 38-CD38
CD38 is an ectoenzyme involved in TAMs promotion and initiation, which is present on the surface of one-third of the cells [165,166].
The selective inhibition of CD38 with a monoclonal FDA-approved antibody called daratumumab (DARA) enhances tumour immune recognition and reduces tumour growth in vitro and in GBM mouse models. In particular, the association of TMZ and DARA produces better results in terms of anti-tumoral apoptotic effects than TMZ alone [167]. In light of these data, future experiments will be necessary to understand better the role of CD38 in GBM’s microenvironment [168].
5.2.6. Programmed Death Ligand 1-PD-L1
In physiological conditions, the programmed death-1 receptor (PD-1)/Programmed death ligand 1 (PD-L1) axis plays an active role in immune homeostasis and prevents autoimmune response thanks to the activation of Treg cells and the inhibition of aberrant self-reactive T-cells [169,170].
PD-L1 belongs, together with PD-L2, to the B7 proteins family. Both bind to PD-1, a receptor of the CD28/cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) family [171]. The role of this axis as therapeutic target is well known in several solid malignancies [172,173,174].
GBM cells can upregulate the activity of the co-inhibitory pathway B7-CD28 to induce TME immunosuppression and escape from immune control [175].
In fact, PD-L1 is expressed on the surface of GBM cells, tumour infiltrating myeloid cells (TIMs), B-cells, and CNS cells. Recent data suggest that TIMs show the largest amount of PD-L1 molecules, promoted by IL-10 secreted by tumour cells [176,177,178].
The binding of PD-L1 with PD-1 leads to the activation of an immunosuppressive pathway in which the tyrosine phosphatase SHP2 dephosphorylates Zap70, downregulating the cytotoxic activity of lymphocytes and, simultaneously, enhancing the migration ability of GBM cells [176].
The activity of PD-L1 is undoubtedly complex, as shown by the numerous receptors to which it binds, such as PD-1, CD28, CD80 and CTLA-4. However, this mechanism grants to target multiple immune-pathway simultaneously, thanks to the inhibition of only PD-L1 [171].
Instead, PD-L2 is expressed primarily by DCs rather than tumour cells, is stimulated by several TME cytokines, such as IL-15 and IL-7, and binds only to PD-1. This protein is less characterized in GBM than PD-L1, but it has been revealing as a prognostic biomarker. In particular, the overexpression of PD-L2 is related to worse overall survival in GBM patients [171,179].
Conversely, relevant studies also showed how PD-L1 expression in GBM’s microenvironment could have a prognostic impact; in particular, if the expression is high in neurons and low in GBM cells, patients show better outcomes [180]. In contrast, increased expression in glioma cells is related to high tumour grade and worse patient outcomes [171,181].
According to these data, a new therapy based on the inhibition of the signalling cascade mediated by PD-1/PD-L1 has been developed to enhance GBM antigens’ recognition by the self-immune system [182]. Preclinical studies showed a significant regression of tumour mass and a longer survival time in GBM mouse models [176]. In patients with recurrent GBM, monoclonal antibodies against PD-1 and PD-L1 are now being tested within clinical trials. Therapies based on PD-1 inhibitors, such as pembrolizumab and nivolumab, have shown promising results in non-small lung cancer and melanoma but still represent a poor option in GBM due to the peculiar, high immunosuppressive state of this tumour [183,184,185,186]. Similarly, PD-L1 inhibitors, such as durvolumab or atezolizumab, have reported low efficacy in patients with GBM, with only minimal survival improvement for a subgroup of GBM patients with mismatch repair deficiency [187,188]. However, preclinical studies showed that PD-L1 pathway remains immunologically significant, paving the way for new therapeutic strategies [177,178].
5.2.7. Cytotoxic T-Lymphocyte-Associated Protein 4-CTLA-4
CTLA-4 is an immunosuppressive receptor located on the surface of T-cells, which blocks the initiation of the adaptative immune response, binding to specific costimulatory molecules (CD80 and CD86) expressed by APCs and essential for the naïve and memory T-cell activation. Also, the CTLA-4 is involved in the inhibition of T-cells by Tregs to preserve peripheral tolerance [189].
Hence, the inhibition of CTLA-4 unlocks CD4+ and CD8+ effector cells and, at the same time, decreases Tregs activity, supporting and promoting the initiation of an antitumor T-cell response [190,191].
In gliomas, CTLA-4 overexpression is associated with higher tumour grade and worse clinical outcome [192].
To date, ipilimumab, an anti-CTLA-4 monoclonal antibody, has been administered to GBM patients, reporting tolerability troubles [193].
5.2.8. T-Cell Immunoglobulin Mucin-3-TIM-3
T-cell immunoglobulin mucin-3 (TIM3) is a molecule located on the surface of various immune cells, such as Th1 CD4+ and CD8+ cytotoxic T-cells, DCs, NK-cells, and monocytes. The binding of TIM3 with galectin-9, HMGB1 and CEACAM1 leads to immunosuppression and the dysregulation of the innate and adaptative responses.
In cancer, TIM3 may represent an important immunotherapeutic target, particularly in the context of GBM resistance to previous checkpoint inhibition therapies [194,195].
5.2.9. Lymphocyte-Activation Gene 3-LAG-3
Lymphocyte-activation gene 3 (LAG-3) is a molecule presented on the surface of CD4+ and CD8+ T-cells, NK-cells and B-cells [196,197].
LAG3 prevents immune cells’ initiation, binding to crucial receptors, such as MHC-II or LSECtin, to activate CD4+ T-cells or CD8+ T-cell/NK-cells respectively [198].
Studies in humans and mouse models demonstrate that LAG-3 inhibition contrasts GBM progression with particular efficacy in the early stages of treatment [199]. Furthermore, the concomitant synergic blockade of LAG-3 and PD-1 has revealed a more potent anti-tumoral effect than the anti-PD-1 therapy alone in several cancer models [200,201].
These data support the possibility of administering anti-LAG-3 therapy in combination with other checkpoint blockade strategies to achieve a better response in GBM patients.
5.2.10. Cluster of Differentiation 70-CD70
CD70 is a molecule presented on the surface of B- and T-cells, that activates other T- and B- cells or NK-cells, interacting with the glycoprotein CD27 [202].
However, GBM cells may express CD70, promoting immunosuppression through tumour-induced T-cells apoptosis and Tregs’ activation [202,203].
Interestingly, Varlilumab, an anti-CD27 agonist, is now administered in ongoing clinical trials in combination with nivolumab [204].
5.2.11. Gangliosides
Gangliosides are a large family of glycolipids expressed in abundance by GBM cells and represent potential targets for GBM immunotherapy [205].
O-acetylated-GD2 represents the first effective ganglioside target discovered. Indeed, monoclonal antibodies against O-acetylated-GD2, such as Dinutiximab, demonstrated to reduce tumour cells proliferation, both in vivo and in vitro models [206,207]. Moreover, a truncated form of O-acetylated-GD2, delivered by genetically engineered mesenchymal stromal/stem cells, mediates immunoselective recognition of GD2-positive tumours, obtaining a potent anti-tumour activity [208].
6. Cancer Stem Cells and Tumor Microenvironment
Cancer stem cells (CSCs) are involved in tumour initiation, relapse, metastasis and poor survival in multiple malignancies, including GBM [209]. Gliomas contain self-renewing, tumorigenic cells that have been shown to express undifferentiated neural cells markers, such as nestin [31,210,211].
Clonogenic cells with an ambiguous “stem-like” phenotype were found in cortical gliomas bearing p53 mutations. These cells express both neuronal and glial cytoskeletal markers and reveal an epigenetic modulation of anti-apoptotic and de-differentiation genes [31,212,213].
Evidence that tumorigenic stem-like precursor cells resemble glial progenitors, both in markers expression and migratory properties, suggest that glial progenitors that populate the adult CNS represent the source of gliomas [214,215,216].
These cells are capable of self-renewal, proliferation, and differentiation into tumour cells with multiple expression markers, resulting in the building of a heterogeneous tumour [217].
6.1. Glioblastoma’s Molecular Subtypes
Different GBM subtypes are associated with different “cells of origin”, distinguished by peculiar alterations in specific genes, such as EGFR, NF1, and PDGFRA/IDH1 [90,218,219]. Dysregulation of these genes and specific transcription factors (TFs), such as POU3F2, SOX2, SALL2, and OLIG2, drives stem-like cells differentiation into three particular GBM subtypes: the proneural, classical, and mesenchymal [220]. Moreover, different TFs, such as Musashi1 and BIM1, are associated with the expression of the podocalyxin-like protein (PODXL) and the presence of undifferentiated GBM stem-like cells, thus correlating with high glioma grade and worse patients’ outcome [221].
As a result of this heterogeneity, GBM may present a combination of different molecular subtypes within the same tumour in variable percentages, reflecting different responses to therapy: in particular, the classical subtype has the best outcome, whereas the proneural one has the worst [219].
6.2. Glioblastoma Stem Cells’ Niches
GBM CSCs are thought to reside inside microanatomical structures within the tumour (the so-called “tumour niches”), where are maintained and protected from the immune system [222]. Three niches are described within a tumour: the perivascular, the necrotic/hypoxic and the invasive niche. These are functionally distinct, interchangeable, and contain infiltrating myeloid cells, suggesting a close connection and crosstalk between glioma stem cells and the microenvironment [223,224].
These cells play a crucial role in recurrence and disease progression, as demonstrated by their resistance to chemo- and radiotherapy [225,226,227]. Hence, it was recently proposed to target the stem cell-like component of GBM to overcome this issue [228,229,230,231].
GBM CSCs are regulated by intrinsic factors, such as genetic, epigenetic, and metabolic modifications, and by extrinsic factors, including interactions with the TME and the immune system [31].
Transcriptional and epigenetic control, together with several genetic alterations, determine the complex clonal diversity that characterizes GMB [231]. Intertumoral heterogeneity in GBM results from distinct differentiation lineages of different precursor cells, suggesting the presence of multiple stem cell-like populations [219,231].
Similarly, metabolism affects heterogeneity in GBM; indeed, the hypoxic tumour microenvironment supports CSCs self-renewal, proliferation, and survival. IDH1 mutations represent an interesting link between genetics and metabolism in GBM, causing accumulation of 2-hydroxyglutarate, thus promoting aberrant hypermethylation of DNA and histones, and dedifferentiation [232,233,234,235,236,237,238].
CSCs niches result from the complex microenvironmental, metabolic, genetic, and epigenetic alterations that promote tumour growth by activating specific pathways, such as Nf-kB, Notch, BMP and Wnt signalling [234,239,240,241,242,243,244].
CSCs of perivascular niches exploit Notch signalling to interact with the endothelial cells and promote neoangiogenesis through VEGF, supporting infiltrative growth. Moreover, GBM CSCs also play a direct role in building new vessels by transdifferentiation into pericytes [226,245,246,247,248,249].
CSCs stimulate the activation of FoxP3+ regulatory T-cells, which produce IL-10 and TGF-β, that in turn differentiate TAMs into an immunosuppressive M2 phenotype [250,251,252]. This immunosuppressive effect is amplified by the ability of the CSCs to promote the recruitment and attraction of microglia, even more than the differentiated neoplastic cells [222].
Also, CSCs inhibit T-cells proliferation and effector responses, and induce T-cells apoptosis through PD-1, PD-L1, and soluble Galectin-3 [31,250].
These data open future possibilities to combine anti-CSCs drugs with other immunotherapeutic strategies.
7. Future Perspectives
To date, several clinical trials are still ongoing, and different therapeutic approaches against TME have been tested, with alternating achievements. Indeed, improved outcomes were observed only in an extremely selected and very restricted patient population.
The most promising and innovative approaches (other than those directed against GBM tumour cells) are represented by: (i) Varlilumab, an anti-CD27 agonist, administered in combination with Nivolumab [204]; (ii) Daratumumab, an anti-38 agent, already approved for the treatment of multiple myeloma, but still in pre-clinical phase for GBM [168]; (iii) CAR-T cell therapy, even if we are only at the dawn of the application of this innovative technique for GBM treatment [71]; (iv) Dinutiximab, an anti-GD2 agent, previously tested against neuroblastoma [207]; anti-LAG3 (BMS-986016) or anti CD137 (BMS-663513) plus anti-PD1, but still in phase I trial [199]; (v) immune checkpoint inhibitors, such as Nivolumab [253], Pembrolizumab [193,254], Durvalumab [255], Ipilimumab [193] and Atezolizumab [256], alone or in combination with antiangiogenics drugs (i.e., bevacizumab) [257] or dendritic cells vaccines [67,142,143,144,258,259,260]. All these novel therapeutic strategies are summarized in Figure 1.
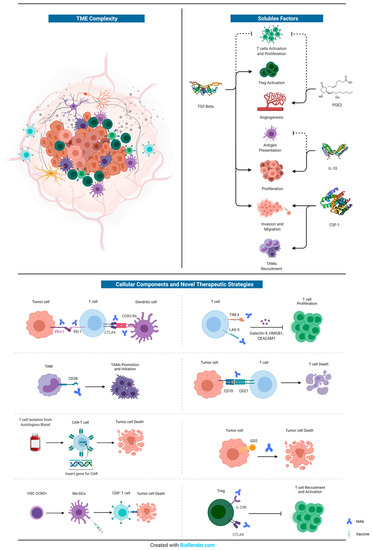
Figure 1.
Schematic representation of the complexity of GBM’s microenvironment. The principal soluble factors with crucial roles in GBM proliferation, migration, neoangiogenesis, and immune-escape are represented in the upper right corner. Instead, a summary of the cellular components of GBM’s immune microenvironment that interact with tumour cells is reported at the bottom, together with novel therapeutic strategies (monoclonal antibodies or vaccines). For further information, see text. MAb: monoclonal antibody.
8. Conclusions
Glioblastoma remains a complex and deadly disease. Deciphering of the multifaced bidirectional network between tumour cells and tumour microenvironment clearly represents the future of translational research, as demonstrated by the several molecular interactions under investigation. To date, some novel targeted approaches are under study; however, a long and winding road is still to be travelled before obtaining a game-changing therapy. Different skills with different viewpoints need to be merged to speed up this process, and a large amount of complex data must be shared within the entire scientific community.
Author Contributions
Conceptualization, C.S., A.G.N. and G.N.F.; data curation, V.O.; writing—original draft preparation, G.N.F. and D.G.; writing—review and editing, F.P., N.M. and P.P.; visualization, C.S. and G.N.F.; supervision, C.S. and A.G.N.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data sharing not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Weller, M.; Cloughesy, T.; Perry, J.R.; Wick, W. Standards of care for treatment of recurrent glioblastoma—Are we there yet? Neuro Oncol. 2013, 15, 4–27. [Google Scholar] [CrossRef]
- Huang, B.; Zhang, H.; Gu, L.; Ye, B.; Jian, Z.; Stary, C.; Xiong, X. Advances in Immunotherapy for Glioblastoma Multiforme. J. Immunol. Res. 2017, 2017, 3597613. [Google Scholar] [CrossRef]
- Ilkhanizadeh, S.; Lau, J.; Huang, M.; Foster, D.J.; Wong, R.; Frantz, A.; Wang, S.; Weiss, W.A.; Persson, A.I. Glial progenitors as targets for transformation in glioma. Adv. Cancer Res. 2014, 121, 1–65. [Google Scholar] [CrossRef]
- Perus, L.J.M.; Walsh, L.A. Microenvironmental Heterogeneity in Brain Malignancies. Front. Immunol. 2019, 10, 2294. [Google Scholar] [CrossRef] [PubMed]
- Taylor, O.G.; Brzozowski, J.S.; Skelding, K.A. Glioblastoma Multiforme: An Overview of Emerging Therapeutic Targets. Front. Oncol. 2019, 9, 963. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Weenink, B.; French, P.J.; Sillevis Smitt, P.A.E.; Debets, R.; Geurts, M. Immunotherapy in Glioblastoma: Current Shortcomings and Future Perspectives. Cancers 2020, 12, 751. [Google Scholar] [CrossRef] [PubMed]
- Karsy, M.; Gelbman, M.; Shah, P.; Balumbu, O.; Moy, F.; Arslan, E. Established and emerging variants of glioblastoma multiforme: Review of morphological and molecular features. Folia Neuropathol. 2012, 50, 301–321. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15017. [Google Scholar] [CrossRef] [PubMed]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee Sh, U. Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Walid, M.S. Prognostic factors for long-term survival after glioblastoma. Perm J. 2008, 12, 45–48. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Kesari, S. Malignant gliomas in adults. N. Engl. J. Med. 2008, 359, 492–507. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Walentynowicz, K.A.; Rajan, W.D.; Kaminska, B. Immune microenvironment of gliomas. Lab. Invest. 2017, 97, 498–518. [Google Scholar] [CrossRef] [PubMed]
- Weller, M.; Stupp, R.; Hegi, M.E.; van den Bent, M.; Tonn, J.C.; Sanson, M.; Wick, W.; Reifenberger, G. Personalized care in neuro-oncology coming of age: Why we need MGMT and 1p/19q testing for malignant glioma patients in clinical practice. Neuro Oncol. 2012, 14 (Suppl. 4), iv100–iv108. [Google Scholar] [CrossRef]
- Lim, M.; Xia, Y.; Bettegowda, C.; Weller, M. Current state of immunotherapy for glioblastoma. Nat. Rev. Clin. Oncol 2018, 15, 422–442. [Google Scholar] [CrossRef] [PubMed]
- Turtoi, A.; Musmeci, D.; Naccarato, A.G.; Scatena, C.; Ortenzi, V.; Kiss, R.; Murtas, D.; Patsos, G.; Mazzucchelli, G.; De Pauw, E.; et al. Sparc-like protein 1 is a new marker of human glioma progression. J. Proteome Res. 2012, 11, 5011–5021. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Goldmann, J.; Kwidzinski, E.; Brandt, C.; Mahlo, J.; Richter, D.; Bechmann, I. T cells traffic from brain to cervical lymph nodes via the cribroid plate and the nasal mucosa. J. Leukoc. Biol. 2006, 80, 797–801. [Google Scholar] [CrossRef]
- Hatterer, E.; Davoust, N.; Didier-Bazes, M.; Vuaillat, C.; Malcus, C.; Belin, M.F.; Nataf, S. How to drain without lymphatics? Dendritic cells migrate from the cerebrospinal fluid to the B-cell follicles of cervical lymph nodes. Blood 2006, 107, 806–812. [Google Scholar] [CrossRef]
- Gowans, J.L.; Knight, E.J. The Route of Re-Circulation of Lymphocytes in the Rat. Proc. R. Soc. Lond. B Biol. Sci. 1964, 159, 257–282. [Google Scholar] [CrossRef] [PubMed]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Iliff, J.J.; Wang, M.; Liao, Y.; Plogg, B.A.; Peng, W.; Gundersen, G.A.; Benveniste, H.; Vates, G.E.; Deane, R.; Goldman, S.A.; et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 2012, 4, 147ra111. [Google Scholar] [CrossRef]
- Louveau, A.; Plog, B.A.; Antila, S.; Alitalo, K.; Nedergaard, M.; Kipnis, J. Understanding the functions and relationships of the glymphatic system and meningeal lymphatics. J. Clin. Investig. 2017, 127, 3210–3219. [Google Scholar] [CrossRef]
- Mestre, H.; Du, T.; Sweeney, A.M.; Liu, G.; Samson, A.J.; Peng, W.; Mortensen, K.N.; Staeger, F.F.; Bork, P.A.R.; Bashford, L.; et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 2020, 367. [Google Scholar] [CrossRef]
- Tamura, R.; Yoshida, K.; Toda, M. Current understanding of lymphatic vessels in the central nervous system. Neurosurg. Rev. 2020, 43, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Cserr, H.F.; Knopf, P.M. Cervical lymphatics, the blood-brain barrier and the immunoreactivity of the brain: A new view. Immunol. Today 1992, 13, 507–512. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Engelhardt, B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat. Rev. Immunol. 2012, 12, 623–635. [Google Scholar] [CrossRef]
- Abels, E.R.; Maas, S.L.N.; Tai, E.; Ting, D.T.; Broekman, M.L.D.; Breakefield, X.O.; El Khoury, J. GlioM&M: Web-based tool for studying circulating and infiltrating monocytes and macrophages in glioma. Sci. Rep. 2020, 10, 9898. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [PubMed]
- Locarno, C.V.; Simonelli, M.; Carenza, C.; Capucetti, A.; Stanzani, E.; Lorenzi, E.; Persico, P.; Della Bella, S.; Passoni, L.; Mavilio, D.; et al. Role of myeloid cells in the immunosuppressive microenvironment in gliomas. Immunobiology 2020, 225, 151853. [Google Scholar] [CrossRef]
- Brown, N.F.; Carter, T.J.; Ottaviani, D.; Mulholland, P. Harnessing the immune system in glioblastoma. Br. J. Cancer 2018, 119, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Gutmann, D.H.; Kettenmann, H. The role of microglia and macrophages in glioma maintenance and progression. Nat. Neurosci. 2016, 19, 20–27. [Google Scholar] [CrossRef]
- Sevenich, L. Brain-Resident Microglia and Blood-Borne Macrophages Orchestrate Central Nervous System Inflammation in Neurodegenerative Disorders and Brain Cancer. Front. Immunol 2018, 9, 697. [Google Scholar] [CrossRef]
- Glass, R.; Synowitz, M. CNS macrophages and peripheral myeloid cells in brain tumours. Acta Neuropathol 2014, 128, 347–362. [Google Scholar] [CrossRef] [PubMed]
- Rajani, K.R.; Carlstrom, L.P.; Parney, I.F.; Johnson, A.J.; Warrington, A.E.; Burns, T.C. Harnessing radiation biology to augment immunotherapy for glioblastoma. Front. Oncol. 2019, 9, 656. [Google Scholar] [CrossRef] [PubMed]
- Razavi, S.M.; Lee, K.E.; Jin, B.E.; Aujla, P.S.; Gholamin, S.; Li, G. Immune Evasion Strategies of Glioblastoma. Front. Surg. 2016, 3, 11. [Google Scholar] [CrossRef]
- Chow, B.W.; Gu, C. The molecular constituents of the blood-brain barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Bechmann, I.; Steiner, B.; Gimsa, U.; Mor, G.; Wolf, S.; Beyer, M.; Nitsch, R.; Zipp, F. Astrocyte-induced T cell elimination is CD95 ligand dependent. J. Neuroimmunol. 2002, 132, 60–65. [Google Scholar] [CrossRef]
- Gold, R.; Schmied, M.; Tontsch, U.; Hartung, H.P.; Wekerle, H.; Toyka, K.V.; Lassmann, H. Antigen presentation by astrocytes primes rat T lymphocytes for apoptotic cell death. A model for T-cell apoptosis in vivo. Brain 1996, 119 Pt 2, 651–659. [Google Scholar] [CrossRef][Green Version]
- Engelhardt, B. The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr. Pharm. Des. 2008, 14, 1555–1565. [Google Scholar] [CrossRef]
- Kmiecik, J.; Poli, A.; Brons, N.H.; Waha, A.; Eide, G.E.; Enger, P.O.; Zimmer, J.; Chekenya, M. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J. Neuroimmunol. 2013, 264, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Woroniecka, K.; Chongsathidkiet, P.; Rhodin, K.; Kemeny, H.; Dechant, C.; Farber, S.H.; Elsamadicy, A.A.; Cui, X.; Koyama, S.; Jackson, C.; et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin. Cancer Res. 2018, 24, 4175–4186. [Google Scholar] [CrossRef] [PubMed]
- Lohr, J.; Ratliff, T.; Huppertz, A.; Ge, Y.; Dictus, C.; Ahmadi, R.; Grau, S.; Hiraoka, N.; Eckstein, V.; Ecker, R.C.; et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin. Cancer Res. 2011, 17, 4296–4308. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Han, Y.; Lesniak, M.S. Prolongation of survival following depletion of CD4+CD25+ regulatory T cells in mice with experimental brain tumors. J. Neurosurg. 2006, 105, 430–437. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Lesniak, M.S. CD4+ CD25+ FoxP3+ T-cell infiltration and heme oxygenase-1 expression correlate with tumor grade in human gliomas. J. Neurooncol. 2007, 83, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Ooi, Y.C.; Tran, P.; Ung, N.; Thill, K.; Trang, A.; Fong, B.M.; Nagasawa, D.T.; Lim, M.; Yang, I. The role of regulatory T-cells in glioma immunology. Clin. Neurol. Neurosurg. 2014, 119, 125–132. [Google Scholar] [CrossRef]
- Adeegbe, D.O.; Nishikawa, H. Natural and induced T regulatory cells in cancer. Front. Immunol. 2013, 4, 190. [Google Scholar] [CrossRef]
- Wainwright, D.A.; Sengupta, S.; Han, Y.; Lesniak, M.S. Thymus-derived rather than tumor-induced regulatory T cells predominate in brain tumors. Neuro Oncol. 2011, 13, 1308–1323. [Google Scholar] [CrossRef] [PubMed]
- Workman, C.J.; Szymczak-Workman, A.L.; Collison, L.W.; Pillai, M.R.; Vignali, D.A. The development and function of regulatory T cells. Cell Mol. Life Sci. 2009, 66, 2603–2622. [Google Scholar] [CrossRef] [PubMed]
- DiDomenico, J.; Lamano, J.B.; Oyon, D.; Li, Y.; Veliceasa, D.; Kaur, G.; Ampie, L.; Choy, W.; Lamano, J.B.; Bloch, O. The immune checkpoint protein PD-L1 induces and maintains regulatory T cells in glioblastoma. Oncoimmunology 2018, 7, e1448329. [Google Scholar] [CrossRef] [PubMed]
- Heimberger, A.B.; Abou-Ghazal, M.; Reina-Ortiz, C.; Yang, D.S.; Sun, W.; Qiao, W.; Hiraoka, N.; Fuller, G.N. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin. Cancer Res. 2008, 14, 5166–5172. [Google Scholar] [CrossRef]
- Maloy, K.J.; Erdmann, I.; Basch, V.; Sierro, S.; Kramps, T.A.; Zinkernagel, R.M.; Oehen, S.; Kundig, T.M. Intralymphatic immunization enhances DNA vaccination. Proc. Natl. Acad. Sci. USA 2001, 98, 3299–3303. [Google Scholar] [CrossRef] [PubMed]
- Dieckmann, D.; Plottner, H.; Berchtold, S.; Berger, T.; Schuler, G. Ex vivo isolation and characterization of CD4(+)CD25(+) T cells with regulatory properties from human blood. J. Exp. Med. 2001, 193, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Mitchell, D.A.; Whitesides, J.F.; Xie, W.; Friedman, A.H.; Archer, G.E.; Herndon, J.E., 2nd; Bigner, D.D.; Dranoff, G.; Sampson, J.H. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006, 66, 3294–3302. [Google Scholar] [CrossRef] [PubMed]
- Fecci, P.E.; Sweeney, A.E.; Grossi, P.M.; Nair, S.K.; Learn, C.A.; Mitchell, D.A.; Cui, X.; Cummings, T.J.; Bigner, D.D.; Gilboa, E.; et al. Systemic anti-CD25 monoclonal antibody administration safely enhances immunity in murine glioma without eliminating regulatory T cells. Clin. Cancer Res. 2006, 12, 4294–4305. [Google Scholar] [CrossRef]
- Martinenaite, E.; Munir Ahmad, S.; Hansen, M.; Met, O.; Westergaard, M.W.; Larsen, S.K.; Klausen, T.W.; Donia, M.; Svane, I.M.; Andersen, M.H. CCL22-specific T Cells: Modulating the immunosuppressive tumor microenvironment. Oncoimmunology 2016, 5, e1238541. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Miska, J.; Wainwright, D.A.; Dey, M.; Rivetta, C.V.; Yu, D.; Kanojia, D.; Pituch, K.C.; Qiao, J.; Pytel, P.; et al. CCL2 Produced by the Glioma Microenvironment Is Essential for the Recruitment of Regulatory T Cells and Myeloid-Derived Suppressor Cells. Cancer Res. 2016, 76, 5671–5682. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef]
- Zhai, L.; Ladomersky, E.; Lauing, K.L.; Wu, M.; Genet, M.; Gritsina, G.; Gyorffy, B.; Brastianos, P.K.; Binder, D.C.; Sosman, J.A.; et al. Infiltrating T Cells Increase IDO1 Expression in Glioblastoma and Contribute to Decreased Patient Survival. Clin. Cancer Res. 2017, 23, 6650–6660. [Google Scholar] [CrossRef]
- Fallarino, F.; Grohmann, U.; Hwang, K.W.; Orabona, C.; Vacca, C.; Bianchi, R.; Belladonna, M.L.; Fioretti, M.C.; Alegre, M.L.; Puccetti, P. Modulation of tryptophan catabolism by regulatory T cells. Nat. Immunol. 2003, 4, 1206–1212. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by kynurenines. Adv. Exp. Med. Biol. 2003, 527, 183–190. [Google Scholar] [CrossRef]
- Banissi, C.; Ghiringhelli, F.; Chen, L.; Carpentier, A.F. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother. 2009, 58, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Samson, A.; Scott, K.J.; Taggart, D.; West, E.J.; Wilson, E.; Nuovo, G.J.; Thomson, S.; Corns, R.; Mathew, R.K.; Fuller, M.J.; et al. Intravenous delivery of oncolytic reovirus to brain tumor patients immunologically primes for subsequent checkpoint blockade. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef]
- Jordan, K.R.; McMahan, R.H.; Kemmler, C.B.; Kappler, J.W.; Slansky, J.E. Peptide vaccines prevent tumor growth by activating T cells that respond to native tumor antigens. Proc. Natl. Acad. Sci. USA 2010, 107, 4652–4657. [Google Scholar] [CrossRef]
- Davoodzadeh Gholami, M.; Kardar, G.A.; Saeedi, Y.; Heydari, S.; Garssen, J.; Falak, R. Exhaustion of T lymphocytes in the tumor microenvironment: Significance and effective mechanisms. Cell Immunol. 2017, 322, 1–14. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [PubMed]
- Bagley, S.J.; Desai, A.S.; Linette, G.P.; June, C.H.; O’Rourke, D.M. CAR T-cell therapy for glioblastoma: Recent clinical advances and future challenges. Neuro Oncol. 2018, 20, 1429–1438. [Google Scholar] [CrossRef]
- Migliorini, D.; Dietrich, P.Y.; Stupp, R.; Linette, G.P.; Posey, A.D., Jr.; June, C.H. CAR T-Cell Therapies in Glioblastoma: A First Look. Clin. Cancer Res. 2018, 24, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Hodges, T.R.; Ott, M.; Xiu, J.; Gatalica, Z.; Swensen, J.; Zhou, S.; Huse, J.T.; de Groot, J.; Li, S.; Overwijk, W.W.; et al. Mutational burden, immune checkpoint expression, and mismatch repair in glioma: Implications for immune checkpoint immunotherapy. Neuro Oncol. 2017, 19, 1047–1057. [Google Scholar] [CrossRef]
- Gomez, G.G.; Kruse, C.A. Mechanisms of malignant glioma immune resistance and sources of immunosuppression. Gene Ther. Mol. Biol. 2006, 10, 133–146. [Google Scholar]
- Leone, P.; Shin, E.C.; Perosa, F.; Vacca, A.; Dammacco, F.; Racanelli, V. MHC class I antigen processing and presenting machinery: Organization, function, and defects in tumor cells. J. Natl. Cancer Inst. 2013, 105, 1172–1187. [Google Scholar] [CrossRef]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Brown, C.E.; Badie, B.; Barish, M.E.; Weng, L.; Ostberg, J.R.; Chang, W.C.; Naranjo, A.; Starr, R.; Wagner, J.; Wright, C.; et al. Bioactivity and Safety of IL13Ralpha2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin. Cancer Res. 2015, 21, 4062–4072. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Brown, C.E.; Starr, R.; Aguilar, B.; Shami, A.F.; Martinez, C.; D’Apuzzo, M.; Barish, M.E.; Forman, S.J.; Jensen, M.C. Stem-like tumor-initiating cells isolated from IL13Ralpha2 expressing gliomas are targeted and killed by IL13-zetakine-redirected T Cells. Clin. Cancer Res. 2012, 18, 2199–2209. [Google Scholar] [CrossRef] [PubMed]
- Bowman, R.L.; Klemm, F.; Akkari, L.; Pyonteck, S.M.; Sevenich, L.; Quail, D.F.; Dhara, S.; Simpson, K.; Gardner, E.E.; Iacobuzio-Donahue, C.A.; et al. Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell Rep. 2016, 17, 2445–2459. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Prinz, M. Origin of microglia: Current concepts and past controversies. Cold Spring Harb. Perspect Biol. 2015, 7, a020537. [Google Scholar] [CrossRef] [PubMed]
- Pinton, L.; Masetto, E.; Vettore, M.; Solito, S.; Magri, S.; D’Andolfi, M.; Del Bianco, P.; Lollo, G.; Benoit, J.P.; Okada, H.; et al. The immune suppressive microenvironment of human gliomas depends on the accumulation of bone marrow-derived macrophages in the center of the lesion. J. Immunother. Cancer 2019, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; Nelson, L.H. Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front. Immunol. 2018, 9, 698. [Google Scholar] [CrossRef]
- Laviron, M.; Boissonnas, A. Ontogeny of Tumor-Associated Macrophages. Front. Immunol. 2019, 10, 1799. [Google Scholar] [CrossRef]
- Movahedi, K.; Van Ginderachter, J.A. The Ontogeny and Microenvironmental Regulation of Tumor-Associated Macrophages. Antioxid Redox Signal. 2016, 25, 775–791. [Google Scholar] [CrossRef] [PubMed]
- Pombo Antunes, A.R.; Scheyltjens, I.; Duerinck, J.; Neyns, B.; Movahedi, K.; Van Ginderachter, J.A. Understanding the glioblastoma immune microenvironment as basis for the development of new immunotherapeutic strategies. Elife 2020, 9, e52176. [Google Scholar] [CrossRef]
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; Coussens, L.M.; Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Hedrick, C.C.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018, 24, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Jiang, Y.C.; Sun, C.K.; Chen, Q.M. Role of the tumor microenvironment in tumor progression and the clinical applications (Review). Oncol. Rep. 2016, 35, 2499–2515. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef]
- Gieryng, A.; Pszczolkowska, D.; Bocian, K.; Dabrowski, M.; Rajan, W.D.; Kloss, M.; Mieczkowski, J.; Kaminska, B. Immune microenvironment of experimental rat C6 gliomas resembles human glioblastomas. Sci. Rep. 2017, 7, 17556. [Google Scholar] [CrossRef]
- Choi, J.; Mai, N.; Jackson, C.; Belcaid, Z.; Lim, M. It takes two: Potential therapies and insights involving microglia and macrophages in glioblastoma. Neuroimmunol. Neuroinflamm. 2018, 5, 42. [Google Scholar] [CrossRef]
- Gutmann, D.H.; Kettenmann, H. Microglia/Brain Macrophages as Central Drivers of Brain Tumor Pathobiology. Neuron 2019, 104, 442–449. [Google Scholar] [CrossRef]
- Schiavoni, G.; Gabriele, L.; Mattei, F. The tumor microenvironment: A pitch for multiple players. Front. Oncol. 2013, 3, 90. [Google Scholar] [CrossRef]
- van den Bossche, W.B.L.; Kleijn, A.; Teunissen, C.E.; Voerman, J.S.A.; Teodosio, C.; Noske, D.P.; van Dongen, J.J.M.; Dirven, C.M.F.; Lamfers, M.L.M. Oncolytic virotherapy in glioblastoma patients induces a tumor macrophage phenotypic shift leading to an altered glioblastoma microenvironment. Neuro Oncol. 2018, 20, 1494–1504. [Google Scholar] [CrossRef] [PubMed]
- Ochocka, N.; Segit, P.; Walentynowicz, K.A.; Wojnicki, K.; Cyranowski, S.; Swatler, J.; Mieczkowski, J.; Kaminska, B. Single-cell RNA sequencing reveals functional heterogeneity of glioma-associated brain macrophages. Nat. Commun. 2021, 12, 1151. [Google Scholar] [CrossRef] [PubMed]
- Greter, M.; Lelios, I.; Croxford, A.L. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front. Immunol. 2015, 6, 249. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef]
- Cattaneo, M. P2Y12 receptors: Structure and function. J. Thromb. Haemost 2015, 13 (Suppl. 1), S10–S16. [Google Scholar] [CrossRef]
- Zhu, C.; Kros, J.M.; van der Weiden, M.; Zheng, P.; Cheng, C.; Mustafa, D.A. Expression site of P2RY12 in residential microglial cells in astrocytomas correlates with M1 and M2 marker expression and tumor grade. Acta Neuropathol. Commun. 2017, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Brune, B.; Dehne, N.; Grossmann, N.; Jung, M.; Namgaladze, D.; Schmid, T.; von Knethen, A.; Weigert, A. Redox control of inflammation in macrophages. Antioxid Redox Signal. 2013, 19, 595–637. [Google Scholar] [CrossRef] [PubMed]
- Piaszyk-Borychowska, A.; Szeles, L.; Csermely, A.; Chiang, H.C.; Wesoly, J.; Lee, C.K.; Nagy, L.; Bluyssen, H.A.R. Signal Integration of IFN-I and IFN-II With TLR4 Involves Sequential Recruitment of STAT1-Complexes and NFkappaB to Enhance Pro-inflammatory Transcription. Front. Immunol. 2019, 10, 1253. [Google Scholar] [CrossRef] [PubMed]
- Platanitis, E.; Decker, T. Regulatory Networks Involving STATs, IRFs, and NFkappaB in Inflammation. Front. Immunol. 2018, 9, 2542. [Google Scholar] [CrossRef]
- Mantovani, A.; Biswas, S.K.; Galdiero, M.R.; Sica, A.; Locati, M. Macrophage plasticity and polarization in tissue repair and remodelling. J. Pathol. 2013, 229, 176–185. [Google Scholar] [CrossRef]
- Soldano, S.; Pizzorni, C.; Paolino, S.; Trombetta, A.C.; Montagna, P.; Brizzolara, R.; Ruaro, B.; Sulli, A.; Cutolo, M. Alternatively Activated (M2) Macrophage Phenotype Is Inducible by Endothelin-1 in Cultured Human Macrophages. PLoS ONE 2016, 11, e0166433. [Google Scholar] [CrossRef]
- Komai, T.; Inoue, M.; Okamura, T.; Morita, K.; Iwasaki, Y.; Sumitomo, S.; Shoda, H.; Yamamoto, K.; Fujio, K. Transforming Growth Factor-beta and Interleukin-10 Synergistically Regulate Humoral Immunity via Modulating Metabolic Signals. Front. Immunol. 2018, 9, 1364. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Pelz, A.; Feng, X.; Synowitz, M.; Markovic, D.; Langmann, T.; Holtman, I.R.; Wang, X.; Eggen, B.J.; Boddeke, H.W.; et al. Glioma-associated microglia/macrophages display an expression profile different from M1 and M2 polarization and highly express Gpnmb and Spp1. PLoS ONE 2015, 10, e0116644. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, H.; Wang, X.; Jiang, G.; Liu, H.; Zhang, G.; Wang, H.; Fang, R.; Bu, X.; Cai, S.; et al. TGF-beta induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget 2016, 7, 52294–52306. [Google Scholar] [CrossRef] [PubMed]
- Martikainen, M.; Essand, M. Virus-Based Immunotherapy of Glioblastoma. Cancers 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed]
- Aurelian, L. Oncolytic viruses as immunotherapy: Progress and remaining challenges. Oncol. Targets Ther. 2016, 9, 2627–2637. [Google Scholar] [CrossRef]
- Russell, L.; Peng, K.W. The emerging role of oncolytic virus therapy against cancer. Chin. Clin. Oncol. 2018, 7, 16. [Google Scholar] [CrossRef]
- Koks, C.A.; De Vleeschouwer, S.; Graf, N.; Van Gool, S.W. Immune Suppression during Oncolytic Virotherapy for High-Grade Glioma; Yes or No? J. Cancer 2015, 6, 203–217. [Google Scholar] [CrossRef][Green Version]
- Geletneky, K.; Hajda, J.; Angelova, A.L.; Leuchs, B.; Capper, D.; Bartsch, A.J.; Neumann, J.O.; Schoning, T.; Husing, J.; Beelte, B.; et al. Oncolytic H-1 Parvovirus Shows Safety and Signs of Immunogenic Activity in a First Phase I/IIa Glioblastoma Trial. Mol. Ther. 2017, 25, 2620–2634. [Google Scholar] [CrossRef] [PubMed]
- Steinman, R.M.; Banchereau, J. Taking dendritic cells into medicine. Nature 2007, 449, 419–426. [Google Scholar] [CrossRef]
- Gabrilovich, D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nat. Rev. Immunol. 2004, 4, 941–952. [Google Scholar] [CrossRef]
- Lee, J.H.; Choi, S.Y.; Jung, N.C.; Song, J.Y.; Seo, H.G.; Lee, H.S.; Lim, D.S. The Effect of the Tumor Microenvironment and Tumor-Derived Metabolites on Dendritic Cell Function. J. Cancer 2020, 11, 769–775. [Google Scholar] [CrossRef]
- Vicari, A.P.; Caux, C.; Trinchieri, G. Tumour escape from immune surveillance through dendritic cell inactivation. Semin. Cancer Biol. 2002, 12, 33–42. [Google Scholar] [CrossRef]
- Zhu, S.; Yang, N.; Wu, J.; Wang, X.; Wang, W.; Liu, Y.J.; Chen, J. Tumor microenvironment-related dendritic cell deficiency: A target to enhance tumor immunotherapy. Pharmacol. Res. 2020, 159, 104980. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, Q.; Gabrusiewicz, K.; Kong, L.Y.; Xia, X.; Wang, J.; Ott, M.; Xu, J.; Davis, R.E.; Huo, L.; et al. Author Correction: FGL2 promotes tumor progression in the CNS by suppressing CD103(+) dendritic cell differentiation. Nat. Commun. 2019, 10, 862. [Google Scholar] [CrossRef]
- Zhan, Y.; Carrington, E.M.; van Nieuwenhuijze, A.; Bedoui, S.; Seah, S.; Xu, Y.; Wang, N.; Mintern, J.D.; Villadangos, J.A.; Wicks, I.P.; et al. GM-CSF increases cross-presentation and CD103 expression by mouse CD8(+) spleen dendritic cells. Eur. J. Immunol. 2011, 41, 2585–2595. [Google Scholar] [CrossRef]
- Scarlett, U.K.; Rutkowski, M.R.; Rauwerdink, A.M.; Fields, J.; Escovar-Fadul, X.; Baird, J.; Cubillos-Ruiz, J.R.; Jacobs, A.C.; Gonzalez, J.L.; Weaver, J.; et al. Ovarian cancer progression is controlled by phenotypic changes in dendritic cells. J. Exp. Med. 2012, 209, 495–506. [Google Scholar] [CrossRef]
- Laoui, D.; Keirsse, J.; Morias, Y.; Van Overmeire, E.; Geeraerts, X.; Elkrim, Y.; Kiss, M.; Bolli, E.; Lahmar, Q.; Sichien, D.; et al. The tumour microenvironment harbours ontogenically distinct dendritic cell populations with opposing effects on tumour immunity. Nat. Commun. 2016, 7, 13720. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Aymeric, L.; Locher, C.; Kroemer, G.; Zitvogel, L. The dendritic cell-tumor cross-talk in cancer. Curr. Opin. Immunol. 2011, 23, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Gulubova, M. Myeloid and Plasmacytoid Dendritic Cells and Cancer—New Insights. Open Access Maced. J. Med. Sci. 2019, 7, 3324–3340. [Google Scholar] [CrossRef] [PubMed]
- Hubert, M.; Gobbini, E.; Bendriss-Vermare, N.; Caux, C.; Valladeau-Guilemond, J. Human Tumor-Infiltrating Dendritic Cells: From in Situ Visualization to High-Dimensional Analyses. Cancers 2019, 11, 1082. [Google Scholar] [CrossRef]
- Guilliams, M.; Ginhoux, F.; Jakubzick, C.; Naik, S.H.; Onai, N.; Schraml, B.U.; Segura, E.; Tussiwand, R.; Yona, S. Dendritic cells, monocytes and macrophages: A unified nomenclature based on ontogeny. Nat. Rev. Immunol. 2014, 14, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.W.; Broz, M.L.; Binnewies, M.; Headley, M.B.; Nelson, A.E.; Wolf, D.M.; Kaisho, T.; Bogunovic, D.; Bhardwaj, N.; Krummel, M.F. Critical Role for CD103(+)/CD141(+) Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell 2016, 30, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Barry, K.C.; Hsu, J.; Broz, M.L.; Cueto, F.J.; Binnewies, M.; Combes, A.J.; Nelson, A.E.; Loo, K.; Kumar, R.; Rosenblum, M.D.; et al. A natural killer-dendritic cell axis defines checkpoint therapy-responsive tumor microenvironments. Nat. Med. 2018, 24, 1178–1191. [Google Scholar] [CrossRef]
- Bottcher, J.P.; Bonavita, E.; Chakravarty, P.; Blees, H.; Cabeza-Cabrerizo, M.; Sammicheli, S.; Rogers, N.C.; Sahai, E.; Zelenay, S.; Reis, E.S.C. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell 2018, 172, 1022–1037.e1014. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Nish, S.A.; Jiang, R.; Hou, L.; Licona-Limon, P.; Weinstein, J.S.; Zhao, H.; Medzhitov, R. Control of T helper 2 responses by transcription factor IRF4-dependent dendritic cells. Immunity 2013, 39, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Sathe, P.; Helft, J.; Miller, J.; Mortha, A. The dendritic cell lineage: Ontogeny and function of dendritic cells and their subsets in the steady state and the inflamed setting. Annu. Rev. Immunol. 2013, 31, 563–604. [Google Scholar] [CrossRef]
- Soudja, S.M.; Ruiz, A.L.; Marie, J.C.; Lauvau, G. Inflammatory monocytes activate memory CD8(+) T and innate NK lymphocytes independent of cognate antigen during microbial pathogen invasion. Immunity 2012, 37, 549–562. [Google Scholar] [CrossRef]
- Flores, C.T.; Wildes, T.J.; Drake, J.A.; Moore, G.L.; Dean, B.D.; Abraham, R.S.; Mitchell, D.A. Lin(-)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade. Nat. Commun. 2018, 9, 4313. [Google Scholar] [CrossRef]
- Garzon-Muvdi, T.; Theodros, D.; Luksik, A.S.; Maxwell, R.; Kim, E.; Jackson, C.M.; Belcaid, Z.; Ganguly, S.; Tyler, B.; Brem, H.; et al. Dendritic cell activation enhances anti-PD-1 mediated immunotherapy against glioblastoma. Oncotarget 2018, 9, 20681–20697. [Google Scholar] [CrossRef]
- Miao, X.; Chen, Y.; Hao, K.; Zheng, M.; Chen, B.; Li, K.; Wang, Y.; Zhang, W.; Zhang, Y.; Mou, X.; et al. CD103(+) Cell Growth Factor Flt3L Enhances the Efficacy of Immune Checkpoint Blockades in Murine Glioblastoma Model. Oncol. Res. 2018, 26, 173–182. [Google Scholar] [CrossRef]
- Wang, J.; Liu, P.; Xin, S.; Wang, Z.; Li, J. Nrf2 suppresses the function of dendritic cells to facilitate the immune escape of glioma cells. Exp. Cell Res. 2017, 360, 66–73. [Google Scholar] [CrossRef]
- Xu, L.W.; Chow, K.K.; Lim, M.; Li, G. Current vaccine trials in glioblastoma: A review. J. Immunol. Res. 2014, 2014, 796856. [Google Scholar] [CrossRef]
- Liau, L.M.; Prins, R.M.; Kiertscher, S.M.; Odesa, S.K.; Kremen, T.J.; Giovannone, A.J.; Lin, J.W.; Chute, D.J.; Mischel, P.S.; Cloughesy, T.F.; et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin. Cancer Res. 2005, 11, 5515–5525. [Google Scholar] [CrossRef]
- Fadul, C.E.; Fisher, J.L.; Hampton, T.H.; Lallana, E.C.; Li, Z.; Gui, J.; Szczepiorkowski, Z.M.; Tosteson, T.D.; Rhodes, C.H.; Wishart, H.A.; et al. Immune response in patients with newly diagnosed glioblastoma multiforme treated with intranodal autologous tumor lysate-dendritic cell vaccination after radiation chemotherapy. J. Immunother. 2011, 34, 382–389. [Google Scholar] [CrossRef]
- Phuphanich, S.; Wheeler, C.J.; Rudnick, J.D.; Mazer, M.; Wang, H.; Nuno, M.A.; Richardson, J.E.; Fan, X.; Ji, J.; Chu, R.M.; et al. Phase I trial of a multi-epitope-pulsed dendritic cell vaccine for patients with newly diagnosed glioblastoma. Cancer Immunol. Immunother. 2013, 62, 125–135. [Google Scholar] [CrossRef]
- Albulescu, R.; Codrici, E.; Popescu, I.D.; Mihai, S.; Necula, L.G.; Petrescu, D.; Teodoru, M.; Tanase, C.P. Cytokine patterns in brain tumour progression. Mediat. Inflamm. 2013, 2013, 979748. [Google Scholar] [CrossRef] [PubMed]
- Sheng, K.C.; Wright, M.D.; Apostolopoulos, V. Inflammatory mediators hold the key to dendritic cell suppression and tumor progression. Curr. Med. Chem. 2011, 18, 5507–5518. [Google Scholar] [CrossRef] [PubMed]
- Couldwell, W.T.; Dore-Duffy, P.; Apuzzo, M.L.; Antel, J.P. Malignant glioma modulation of immune function: Relative contribution of different soluble factors. J. Neuroimmunol. 1991, 33, 89–96. [Google Scholar] [CrossRef]
- Wei, J.; Gabrusiewicz, K.; Heimberger, A. The controversial role of microglia in malignant gliomas. Clin. Dev. Immunol. 2013, 2013, 285246. [Google Scholar] [CrossRef]
- Takenaka, M.C.; Gabriely, G.; Rothhammer, V.; Mascanfroni, I.D.; Wheeler, M.A.; Chao, C.C.; Gutierrez-Vazquez, C.; Kenison, J.; Tjon, E.C.; Barroso, A.; et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat. Neurosci. 2019, 22, 729–740. [Google Scholar] [CrossRef] [PubMed]
- Milner, R. Microglial expression of alphavbeta3 and alphavbeta5 integrins is regulated by cytokines and the extracellular matrix: Beta5 integrin null microglia show no defects in adhesion or MMP-9 expression on vitronectin. Glia 2009, 57, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Wai, P.Y.; Kuo, P.C. The role of Osteopontin in tumor metastasis. J. Surg Res. 2004, 121, 228–241. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Marisetty, A.; Schrand, B.; Gabrusiewicz, K.; Hashimoto, Y.; Ott, M.; Grami, Z.; Kong, L.Y.; Ling, X.; Caruso, H.; et al. Osteopontin mediates glioblastoma-associated macrophage infiltration and is a potential therapeutic target. J. Clin. Invest. 2019, 129, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Fontana, A.; Bodmer, S.; Frei, K.; Malipiero, U.; Siepl, C. Expression of TGF-β2 in Human Glioblastoma: A Role in Resistance to Immune Rejection? John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2007; pp. 232–241. [Google Scholar] [CrossRef]
- Han, J.; Alvarez-Breckenridge, C.A.; Wang, Q.E.; Yu, J. TGF-β Signaling and Its Targeting for Glioma Treatment; E-Century Publishing Corporation: Madison, WI, USA, 2015; Volume 5, pp. 945–955. [Google Scholar]
- Kirkbride, K.C.; Blobe, G.C. Inhibiting the TGF-beta signalling pathway as a means of cancer immunotherapy. Expert Opin. Biol. Ther. 2003, 3, 251–261. [Google Scholar] [CrossRef]
- Onguru, O.; Gamsizkan, M.; Ulutin, C.; Gunhan, O. Cyclooxygenase-2 (Cox-2) expression and angiogenesis in glioblastoma. Neuropathology 2008, 28, 29–34. [Google Scholar] [CrossRef]
- Chiu, W.T.; Shen, S.C.; Chow, J.M.; Lin, C.W.; Shia, L.T.; Chen, Y.C. Contribution of reactive oxygen species to migration/invasion of human glioblastoma cells U87 via ERK-dependent COX-2/PGE2 activation. Neurobiol. Dis. 2010, 37, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Qiu, J.; Li, Q.; Shi, Z. Prostaglandin E2 Signaling: Alternative Target for Glioblastoma? Cell 2017, 3, 75–78. [Google Scholar] [CrossRef]
- Magana-Maldonado, R.; Chavez-Cortez, E.G.; Olascoaga-Arellano, N.K.; Lopez-Mejia, M.; Maldonado-Leal, F.M.; Sotelo, J.; Pineda, B. Immunological Evasion in Glioblastoma. Biomed. Res. Int. 2016, 2016, 7487313. [Google Scholar] [CrossRef]
- Mittal, S.K.; Roche, P.A. Suppression of Antigen Presentation by IL-10; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; Volume 34, pp. 22–27. [Google Scholar]
- Zhang, Z.; Huang, X.; Li, J.; Fan, H.; Yang, F.; Zhang, R.; Yang, Y.; Feng, S.; He, D.; Sun, W.; et al. Interleukin 10 promotes growth and invasion of glioma cells by up-regulating KPNA 2 in vitro. J. Cancer Res. Ther. 2019, 15, 927. [Google Scholar] [CrossRef] [PubMed]
- Coniglio, S.J.; Eugenin, E.; Dobrenis, K.; Stanley, E.R.; West, B.L.; Symons, M.H.; Segall, J.E. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol. Med. 2012, 18, 519–527. [Google Scholar] [CrossRef]
- Stafford, J.H.; Hirai, T.; Deng, L.; Chernikova, S.B.; Urata, K.; West, B.L.; Brown, J.M. Colony stimulating factor 1 receptor inhibition delays recurrence of glioblastoma after radiation by altering myeloid cell recruitment and polarization. Neuro Oncol. 2016, 18, 797–806. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Hara-Yokoyama, M.; Kukimoto-Niino, M.; Terasawa, K.; Harumiya, S.; Podyma-Inoue, K.A.; Hino, N.; Sakamoto, K.; Itoh, S.; Hashii, N.; Hiruta, Y.; et al. Tetrameric interaction of the ectoenzyme CD38 on the cell surface enables its catalytic and raft-association activities. Structure 2012, 20, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Cuzzubbo, S.; McArthur, S.; Durrant, L.G.; Adhikaree, J.; Tinsley, C.J.; Pockley, A.G.; McArdle, S.E.B. Immune Escape in Glioblastoma Multiforme and the Adaptation of Immunotherapies for Treatment. Front. Immunol. 2020, 11, 582106. [Google Scholar] [CrossRef] [PubMed]
- Blacher, E.; Ben Baruch, B.; Levy, A.; Geva, N.; Green, K.D.; Garneau-Tsodikova, S.; Fridman, M.; Stein, R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer 2015, 136, 1422–1433. [Google Scholar] [CrossRef]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 pathway in tolerance and autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef]
- Chen, R.Q.; Liu, F.; Qiu, X.Y.; Chen, X.Q. The Prognostic and Therapeutic Value of PD-L1 in Glioma. Front. Pharmacol. 2018, 9, 1503. [Google Scholar] [CrossRef] [PubMed]
- Akinleye, A.; Rasool, Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J. Hematol. Oncol. 2019, 12, 92. [Google Scholar] [CrossRef]
- Saraggi, D.; Galuppini, F.; Remo, A.; Urso, E.D.L.; Bacchin, D.; Salmaso, R.; Lanza, C.; Bao, R.Q.; Fanelli, G.N.; Guzzardo, V.; et al. PD-L1 overexpression in ampulla of Vater carcinoma and its pre-invasive lesions. Histopathology 2017, 71, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Fassan, M.; Vianello, L.; Sacchi, D.; Fanelli, G.N.; Munari, G.; Scarpa, M.; Cappellesso, R.; Loupakis, F.; Lanza, C.; Salmaso, R.; et al. Correction to: Assessment of intratumor immune-microenvironment in colorectal cancers with extranodal extension of nodal metastases. Cancer Cell Int. 2019, 19, 244. [Google Scholar] [CrossRef] [PubMed]
- Schildberg, F.A.; Klein, S.R.; Freeman, G.J.; Sharpe, A.H. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity 2016, 44, 955–972. [Google Scholar] [CrossRef] [PubMed]
- Litak, J.; Mazurek, M.; Grochowski, C.; Kamieniak, P.; Rolinski, J. PD-L1/PD-1 Axis in Glioblastoma Multiforme. Int. J. Mol. Sci. 2019, 20, 5347. [Google Scholar] [CrossRef]
- Antonios, J.P.; Soto, H.; Everson, R.G.; Moughon, D.; Orpilla, J.R.; Shin, N.P.; Sedighim, S.; Treger, J.; Odesa, S.; Tucker, A.; et al. Immunosuppressive tumor-infiltrating myeloid cells mediate adaptive immune resistance via a PD-1/PD-L1 mechanism in glioblastoma. Neuro Oncol. 2017, 19, 796–807. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Crane, C.A.; Kaur, R.; Safaee, M.; Rutkowski, M.J.; Parsa, A.T. Gliomas promote immunosuppression through induction of B7-H1 expression in tumor-associated macrophages. Clin. Cancer Res. 2013, 19, 3165–3175. [Google Scholar] [CrossRef]
- Wang, Z.L.; Li, G.Z.; Wang, Q.W.; Bao, Z.S.; Wang, Z.; Zhang, C.B.; Jiang, T. PD-L2 expression is correlated with the molecular and clinical features of glioma, and acts as an unfavorable prognostic factor. Oncoimmunology 2019, 8, e1541535. [Google Scholar] [CrossRef]
- Liu, Y.; Carlsson, R.; Ambjorn, M.; Hasan, M.; Badn, W.; Darabi, A.; Siesjo, P.; Issazadeh-Navikas, S. PD-L1 expression by neurons nearby tumors indicates better prognosis in glioblastoma patients. J. Neurosci. 2013, 33, 14231–14245. [Google Scholar] [CrossRef]
- Xue, S.; Song, G.; Yu, J. The prognostic significance of PD-L1 expression in patients with glioma: A meta-analysis. Sci. Rep. 2017, 7, 4231. [Google Scholar] [CrossRef]
- Indraccolo, S.; Lombardi, G.; Fassan, M.; Pasqualini, L.; Giunco, S.; Marcato, R.; Gasparini, A.; Candiotto, C.; Nalio, S.; Fiduccia, P.; et al. Genetic, Epigenetic, and Immunologic Profiling of MMR-Deficient Relapsed Glioblastoma. Clin. Cancer Res. 2019, 25, 1828–1837. [Google Scholar] [CrossRef]
- Santini, F.C.; Hellmann, M.D. PD-1/PD-L1 Axis in Lung Cancer. Cancer J. 2018, 24, 15–19. [Google Scholar] [CrossRef]
- Dang, T.O.; Ogunniyi, A.; Barbee, M.S.; Drilon, A. Pembrolizumab for the treatment of PD-L1 positive advanced or metastatic non-small cell lung cancer. Expert Rev. Anticancer Ther. 2016, 16, 13–20. [Google Scholar] [CrossRef]
- Redman, J.M.; Gibney, G.T.; Atkins, M.B. Advances in immunotherapy for melanoma. BMC Med. 2016, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Filley, A.C.; Henriquez, M.; Dey, M. Recurrent glioma clinical trial, CheckMate-143: The game is not over yet. Oncotarget 2017, 8, 91779–91794. [Google Scholar] [CrossRef] [PubMed]
- Johanns, T.M.; Miller, C.A.; Dorward, I.G.; Tsien, C.; Chang, E.; Perry, A.; Uppaluri, R.; Ferguson, C.; Schmidt, R.E.; Dahiya, S.; et al. Immunogenomics of Hypermutated Glioblastoma: A Patient with Germline POLE Deficiency Treated with Checkpoint Blockade Immunotherapy. Cancer Discov. 2016, 6, 1230–1236. [Google Scholar] [CrossRef]
- Blumenthal, D.T.; Yalon, M.; Vainer, G.W.; Lossos, A.; Yust, S.; Tzach, L.; Cagnano, E.; Limon, D.; Bokstein, F. Pembrolizumab: First experience with recurrent primary central nervous system (CNS) tumors. J. Neurooncol. 2016, 129, 453–460. [Google Scholar] [CrossRef] [PubMed]
- Buchbinder, E.I.; Desai, A. CTLA-4 and PD-1 Pathways: Similarities, Differences, and Implications of Their Inhibition. Am. J. Clin. Oncol. 2016, 39, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Wolchok, J.D.; Saenger, Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist 2008, 13 (Suppl. 4), 2–9. [Google Scholar] [CrossRef] [PubMed]
- Tang, F.; Du, X.; Liu, M.; Zheng, P.; Liu, Y. Anti-CTLA-4 antibodies in cancer immunotherapy: Selective depletion of intratumoral regulatory T cells or checkpoint blockade? Cell Biosci. 2018, 8, 30. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Huang, J.; Liu, X.; Cheng, Q.; Luo, C.; Liu, Z. CTLA-4 correlates with immune and clinical characteristics of glioma. Cancer Cell Int. 2020, 20, 7. [Google Scholar] [CrossRef]
- Omuro, A.; Vlahovic, G.; Lim, M.; Sahebjam, S.; Baehring, J.; Cloughesy, T.; Voloschin, A.; Ramkissoon, S.H.; Ligon, K.L.; Latek, R.; et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: Results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018, 20, 674–686. [Google Scholar] [CrossRef]
- Zhu, C.; Anderson, A.C.; Kuchroo, V.K. TIM-3 and its regulatory role in immune responses. Curr. Top. Microbiol. Immunol. 2011, 350, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Chen, L.; Liu, J.; Yang, L.; Zhang, M.; Wang, L.; Zhang, R.; Uemura, Y.; Wu, Q.; Yu, X.; et al. Current state and future of co-inhibitory immune checkpoints for the treatment of glioblastoma. Cancer Biol. Med. 2020, 17, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Dranoff, G. Cancer Immunology and Immunotherapy; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 2011. [Google Scholar]
- Andrews, L.P.; Marciscano, A.E.; Drake, C.G.; Vignali, D.A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 2017, 276, 80–96. [Google Scholar] [CrossRef]
- Long, L.; Zhang, X.; Chen, F.; Pan, Q.; Phiphatwatchara, P.; Zeng, Y.; Chen, H. The promising immune checkpoint LAG-3: From tumor microenvironment to cancer immunotherapy. Genes Cancer 2018, 9, 176–189. [Google Scholar] [CrossRef]
- Harris-Bookman, S.; Mathios, D.; Martin, A.M.; Xia, Y.; Kim, E.; Xu, H.; Belcaid, Z.; Polanczyk, M.; Barberi, T.; Theodros, D.; et al. Expression of LAG-3 and efficacy of combination treatment with anti-LAG-3 and anti-PD-1 monoclonal antibodies in glioblastoma. Int. J. Cancer 2018, 143, 3201–3208. [Google Scholar] [CrossRef]
- Woo, S.R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef]
- Huang, R.Y.; Eppolito, C.; Lele, S.; Shrikant, P.; Matsuzaki, J.; Odunsi, K. LAG3 and PD1 co-inhibitory molecules collaborate to limit CD8+ T cell signaling and dampen antitumor immunity in a murine ovarian cancer model. Oncotarget 2015, 6, 27359–27377. [Google Scholar] [CrossRef]
- Wischhusen, J.; Jung, G.; Radovanovic, I.; Beier, C.; Steinbach, J.P.; Rimner, A.; Huang, H.; Schulz, J.B.; Ohgaki, H.; Aguzzi, A.; et al. Identification of CD70-mediated apoptosis of immune effector cells as a novel immune escape pathway of human glioblastoma. Cancer Res. 2002, 62, 2592–2599. [Google Scholar]
- Chahlavi, A.; Rayman, P.; Richmond, A.L.; Biswas, K.; Zhang, R.; Vogelbaum, M.; Tannenbaum, C.; Barnett, G.; Finke, J.H. Glioblastomas induce T-lymphocyte death by two distinct pathways involving gangliosides and CD70. Cancer Res. 2005, 65, 5428–5438. [Google Scholar] [CrossRef]
- Starzer, A.M.; Berghoff, A.S. New emerging targets in cancer immunotherapy: CD27 (TNFRSF7). ESMO Open 2020, 4. [Google Scholar] [CrossRef] [PubMed]
- Fleurence, J.; Fougeray, S.; Bahri, M.; Cochonneau, D.; Clemenceau, B.; Paris, F.; Heczey, A.; Birkle, S. Targeting O-Acetyl-GD2 Ganglioside for Cancer Immunotherapy. J. Immunol. Res. 2017, 2017, 5604891. [Google Scholar] [CrossRef]
- Fleurence, J.; Cochonneau, D.; Fougeray, S.; Oliver, L.; Geraldo, F.; Terme, M.; Dorvillius, M.; Loussouarn, D.; Vallette, F.; Paris, F.; et al. Targeting and killing glioblastoma with monoclonal antibody to O-acetyl GD2 ganglioside. Oncotarget 2016, 7, 41172–41185. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.; Wilken, F.; Wagner, I.; Marx, M.; Troschke-Meurer, S.; Zumpe, M.; Bien-Moeller, S.; Weidemeier, M.; Baldauf, J.; Fleck, S.K.; et al. GD2 targeting by dinutuximab beta is a promising immunotherapeutic approach against malignant glioma. J. Neurooncol. 2020, 147, 577–585. [Google Scholar] [CrossRef]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. 2020, 27, 558–570. [Google Scholar] [CrossRef] [PubMed]
- Scatena, C.; Roncella, M.; Di Paolo, A.; Aretini, P.; Menicagli, M.; Fanelli, G.; Marini, C.; Mazzanti, C.M.; Ghilli, M.; Sotgia, F.; et al. Doxycycline, an Inhibitor of Mitochondrial Biogenesis, Effectively Reduces Cancer Stem Cells (CSCs) in Early Breast Cancer Patients: A Clinical Pilot Study. Front. Oncol. 2018, 8, 452. [Google Scholar] [CrossRef] [PubMed]
- Ignatova, T.N.; Kukekov, V.G.; Laywell, E.D.; Suslov, O.N.; Vrionis, F.D.; Steindler, D.A. Human cortical glial tumors contain neural stem-like cells expressing astroglial and neuronal markers in vitro. Glia 2002, 39, 193–206. [Google Scholar] [CrossRef]
- Fanelli, G.N.; Naccarato, A.G.; Scatena, C. Recent Advances in Cancer Plasticity: Cellular Mechanisms, Surveillance Strategies, and Therapeutic Optimization. Front. Oncol. 2020, 10, 569. [Google Scholar] [CrossRef]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res. 2004, 64, 7011–7021. [Google Scholar] [CrossRef]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Canoll, P.; Goldman, J.E. The interface between glial progenitors and gliomas. Acta Neuropathol. 2008, 116, 465–477. [Google Scholar] [CrossRef]
- Caussinus, E.; Gonzalez, C. Induction of tumor growth by altered stem-cell asymmetric division in Drosophila melanogaster. Nat. Genet. 2005, 37, 1125–1129. [Google Scholar] [CrossRef]
- Uhrbom, L.; Kastemar, M.; Johansson, F.K.; Westermark, B.; Holland, E.C. Cell type-specific tumor suppression by Ink4a and Arf in Kras-induced mouse gliomagenesis. Cancer Res. 2005, 65, 2065–2069. [Google Scholar] [CrossRef]
- Hemmati, H.D.; Nakano, I.; Lazareff, J.A.; Masterman-Smith, M.; Geschwind, D.H.; Bronner-Fraser, M.; Kornblum, H.I. Cancerous stem cells can arise from pediatric brain tumors. Proc. Natl. Acad. Sci. USA 2003, 100, 15178–15183. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Verhaak, R.G.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Suva, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014, 157, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Binder, Z.A.; Siu, I.M.; Eberhart, C.G.; Ap Rhys, C.; Bai, R.Y.; Staedtke, V.; Zhang, H.; Smoll, N.R.; Piantadosi, S.; Piccirillo, S.G.; et al. Podocalyxin-like protein is expressed in glioblastoma multiforme stem-like cells and is associated with poor outcome. PLoS ONE 2013, 8, e75945. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, D.; Bergers, G. Glioblastoma: Defining Tumor Niches. Trends Cancer 2015, 1, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Filatova, A.; Acker, T.; Garvalov, B.K. The cancer stem cell niche(s): The crosstalk between glioma stem cells and their microenvironment. Biochim. Biophys. Acta 2013, 1830, 2496–2508. [Google Scholar] [CrossRef]
- Schiffer, D.; Annovazzi, L.; Casalone, C.; Corona, C.; Mellai, M. Glioblastoma: Microenvironment and Niche Concept. Cancers 2018, 11, 5. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, Y.; Yu, T.S.; McKay, R.M.; Burns, D.K.; Kernie, S.G.; Parada, L.F. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature 2012, 488, 522–526. [Google Scholar] [CrossRef]
- Berger, F.; Gay, E.; Pelletier, L.; Tropel, P.; Wion, D. Development of gliomas: Potential role of asymmetrical cell division of neural stem cells. Lancet Oncol. 2004, 5, 511–514. [Google Scholar] [CrossRef]
- Fomchenko, E.I.; Holland, E.C. Stem cells and brain cancer. Exp. Cell Res. 2005, 306, 323–329. [Google Scholar] [CrossRef]
- Oliver, T.G.; Wechsler-Reya, R.J. Getting at the root and stem of brain tumors. Neuron 2004, 42, 885–888. [Google Scholar] [CrossRef] [PubMed]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef]
- Intlekofer, A.M.; Dematteo, R.G.; Venneti, S.; Finley, L.W.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef]
- Kathagen, A.; Schulte, A.; Balcke, G.; Phillips, H.S.; Martens, T.; Matschke, J.; Gunther, H.S.; Soriano, R.; Modrusan, Z.; Sandmann, T.; et al. Hypoxia and oxygenation induce a metabolic switch between pentose phosphate pathway and glycolysis in glioma stem-like cells. Acta Neuropathol. 2013, 126, 763–780. [Google Scholar] [CrossRef]
- Li, Z.; Wang, H.; Eyler, C.E.; Hjelmeland, A.B.; Rich, J.N. Turning cancer stem cells inside out: An exploration of glioma stem cell signaling pathways. J. Biol. Chem. 2009, 284, 16705–16709. [Google Scholar] [CrossRef]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef]
- Schvartzman, J.M.; Reuter, V.P.; Koche, R.P.; Thompson, C.B. 2-hydroxyglutarate inhibits MyoD-mediated differentiation by preventing H3K9 demethylation. Proc. Natl. Acad. Sci. USA 2019, 116, 12851–12856. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062–16067. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, S.; Lessi, F.; Aretini, P.; Ortenzi, V.; Scatena, C.; Menicagli, M.; La Ferla, M.; Civita, P.; Zavaglia, K.; Scopelliti, C.; et al. Cancer astrocytes have a more conserved molecular status in long recurrence free survival (RFS) IDH1 wild-type glioblastoma patients: New emerging cancer players. Oncotarget 2018, 9, 24014–24027. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, R.; Bentivegna, A. Role of Notch Signaling Pathway in Glioblastoma Pathogenesis. Cancers 2019, 11, 292. [Google Scholar] [CrossRef]
- Clark, P.A.; Treisman, D.M.; Ebben, J.; Kuo, J.S. Developmental signaling pathways in brain tumor-derived stem-like cells. Dev. Dyn 2007, 236, 3297–3308. [Google Scholar] [CrossRef] [PubMed]
- Hai, L.; Zhang, C.; Li, T.; Zhou, X.; Liu, B.; Li, S.; Zhu, M.; Lin, Y.; Yu, S.; Zhang, K.; et al. Notch1 is a prognostic factor that is distinctly activated in the classical and proneural subtype of glioblastoma and that promotes glioma cell survival via the NF-kappaB(p65) pathway. Cell Death Dis. 2018, 9, 158. [Google Scholar] [CrossRef] [PubMed]
- Nusblat, L.M.; Tanna, S.; Roth, C.M. Gene silencing of HIF-2alpha disrupts glioblastoma stem cell phenotype. Cancer Drug Resist. 2020, 3, 199–208. [Google Scholar] [CrossRef]
- Rheinbay, E.; Suva, M.L.; Gillespie, S.M.; Wakimoto, H.; Patel, A.P.; Shahid, M.; Oksuz, O.; Rabkin, S.D.; Martuza, R.L.; Rivera, M.N.; et al. An aberrant transcription factor network essential for Wnt signaling and stem cell maintenance in glioblastoma. Cell Rep. 2013, 3, 1567–1579. [Google Scholar] [CrossRef]
- Yan, K.; Wu, Q.; Yan, D.H.; Lee, C.H.; Rahim, N.; Tritschler, I.; DeVecchio, J.; Kalady, M.F.; Hjelmeland, A.B.; Rich, J.N. Glioma cancer stem cells secrete Gremlin1 to promote their maintenance within the tumor hierarchy. Genes Dev. 2014, 28, 1085–1100. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Jhaveri, N.; Chen, T.C.; Hofman, F.M. Tumor vasculature and glioma stem cells: Contributions to glioma progression. Cancer Lett. 2016, 380, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Kristoffersen, K.; Villingshoj, M.; Poulsen, H.S.; Stockhausen, M.T. Level of Notch activation determines the effect on growth and stem cell-like features in glioblastoma multiforme neurosphere cultures. Cancer Biol. Ther. 2013, 14, 625–637. [Google Scholar] [CrossRef]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef]
- Bayin, N.S.; Frenster, J.D.; Sen, R.; Si, S.; Modrek, A.S.; Galifianakis, N.; Dolgalev, I.; Ortenzi, V.; Illa-Bochaca, I.; Khahera, A.; et al. Notch signaling regulates metabolic heterogeneity in glioblastoma stem cells. Oncotarget 2017, 8, 64932–64953. [Google Scholar] [CrossRef]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M.; et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Ye, H.; Qi, Z.; Mo, L.; Yue, Q.; Baral, A.; Hoon, D.S.B.; Vera, J.C.; Heiss, J.D.; Chen, C.C.; et al. B7-H4(B7x)-Mediated Cross-talk between Glioma-Initiating Cells and Macrophages via the IL6/JAK/STAT3 Pathway Lead to Poor Prognosis in Glioma Patients. Clin. Cancer Res. 2016, 22, 2778–2790. [Google Scholar] [CrossRef]
- Schalper, K.A.; Rodriguez-Ruiz, M.E.; Diez-Valle, R.; Lopez-Janeiro, A.; Porciuncula, A.; Idoate, M.A.; Inoges, S.; de Andrea, C.; Lopez-Diaz de Cerio, A.; Tejada, S.; et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat. Med. 2019, 25, 470–476. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Mochizuki, A.Y.; Orpilla, J.R.; Hugo, W.; Lee, A.H.; Davidson, T.B.; Wang, A.C.; Ellingson, B.M.; Rytlewski, J.A.; Sanders, C.M.; et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat. Med. 2019, 25, 477–486. [Google Scholar] [CrossRef]
- Sheth, S.; Gao, C.; Mueller, N.; Angra, N.; Gupta, A.; Germa, C.; Martinez, P.; Soria, J.C. Durvalumab activity in previously treated patients who stopped durvalumab without disease progression. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Lukas, R.V.; Rodon, J.; Becker, K.; Wong, E.T.; Shih, K.; Touat, M.; Fasso, M.; Osborne, S.; Molinero, L.; O’Hear, C.; et al. Clinical activity and safety of atezolizumab in patients with recurrent glioblastoma. J. Neurooncol. 2018, 140, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Nayak, L.; Peters, K.B.; Clarke, J.L.; Jordan, J.T.; De Groot, J.F.; Nghiemphu, P.L.; Kaley, T.J.; Colman, H.; Gaffey, S.C.; et al. Phase II study of pembrolizumab or pembrolizumab plus bevacizumab for recurrent glioblastoma (rGBM) patients. J. Clin. Oncol. 2018, 36, 2006. [Google Scholar] [CrossRef]
- Liau, L.M.; Ashkan, K.; Tran, D.D.; Campian, J.L.; Trusheim, J.E.; Cobbs, C.S.; Heth, J.A.; Salacz, M.; Taylor, S.; D’Andre, S.D.; et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J. Transl. Med. 2018, 16, 142. [Google Scholar] [CrossRef]
- Chang, C.N.; Huang, Y.C.; Yang, D.M.; Kikuta, K.; Wei, K.J.; Kubota, T.; Yang, W.K. A phase I/II clinical trial investigating the adverse and therapeutic effects of a postoperative autologous dendritic cell tumor vaccine in patients with malignant glioma. J. Clin. Neurosci. 2011, 18, 1048–1054. [Google Scholar] [CrossRef]
- Ardon, H.; Verbinnen, B.; Maes, W.; Beez, T.; Van Gool, S.; De Vleeschouwer, S. Technical advancement in regulatory T cell isolation and characterization using CD127 expression in patients with malignant glioma treated with autologous dendritic cell vaccination. J. Immunol. Methods 2010, 352, 169–173. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).