
Pilot Study to Detect Genes Involved in DNA Damage and Cancer in Humans: Potential Biomarkers of Exposure to E-Cigarette Aerosols

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Sample Size
2.3. Human Exposure
2.4. Topography Data Collection
2.5. Biologic Sample Collection
2.6. Quantitative RT-PCR Analysis of Gene Expression
2.7. Gene Network Analysis and Biomarker Identification
2.8. Statistical Analysis
3. Results
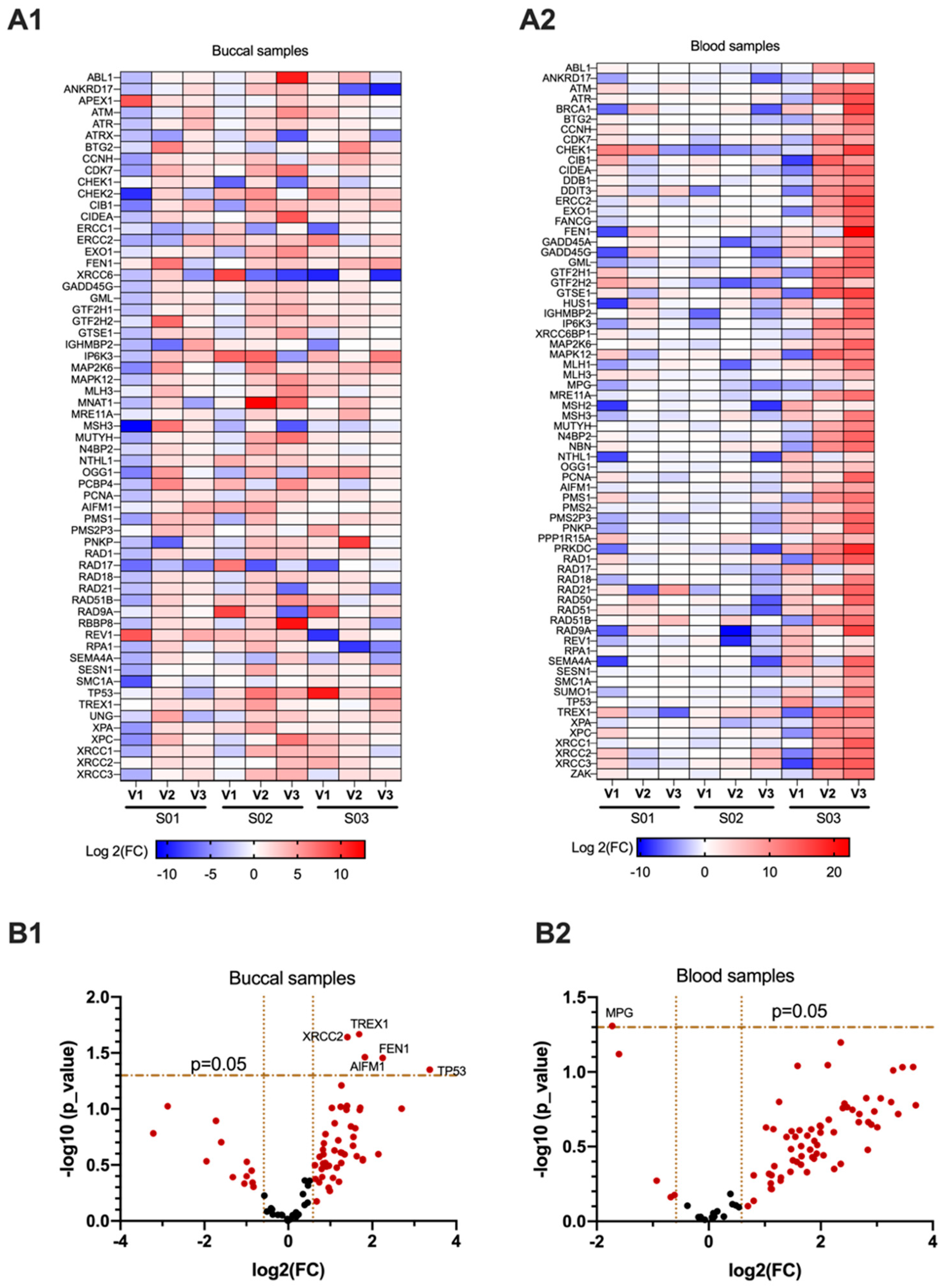
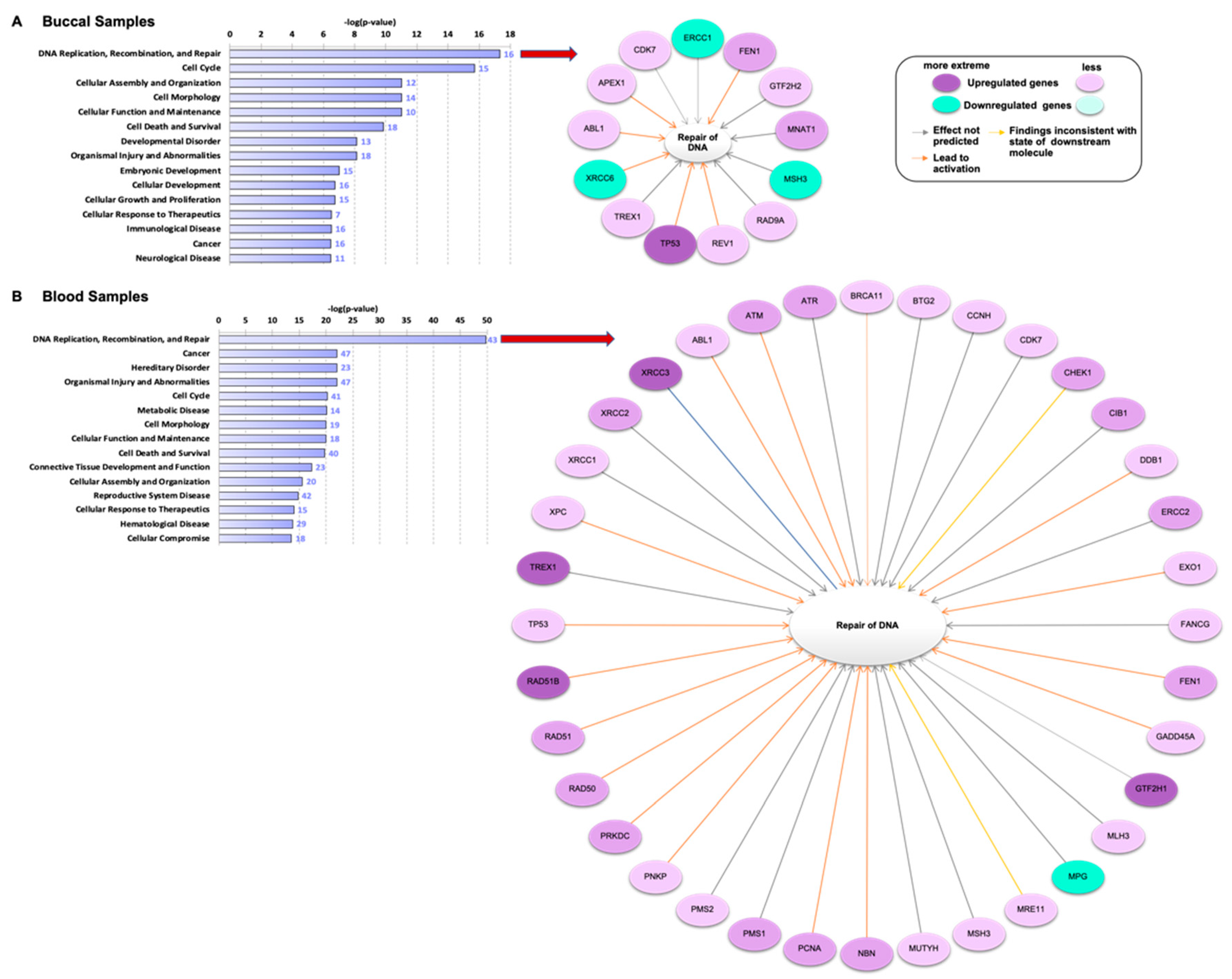
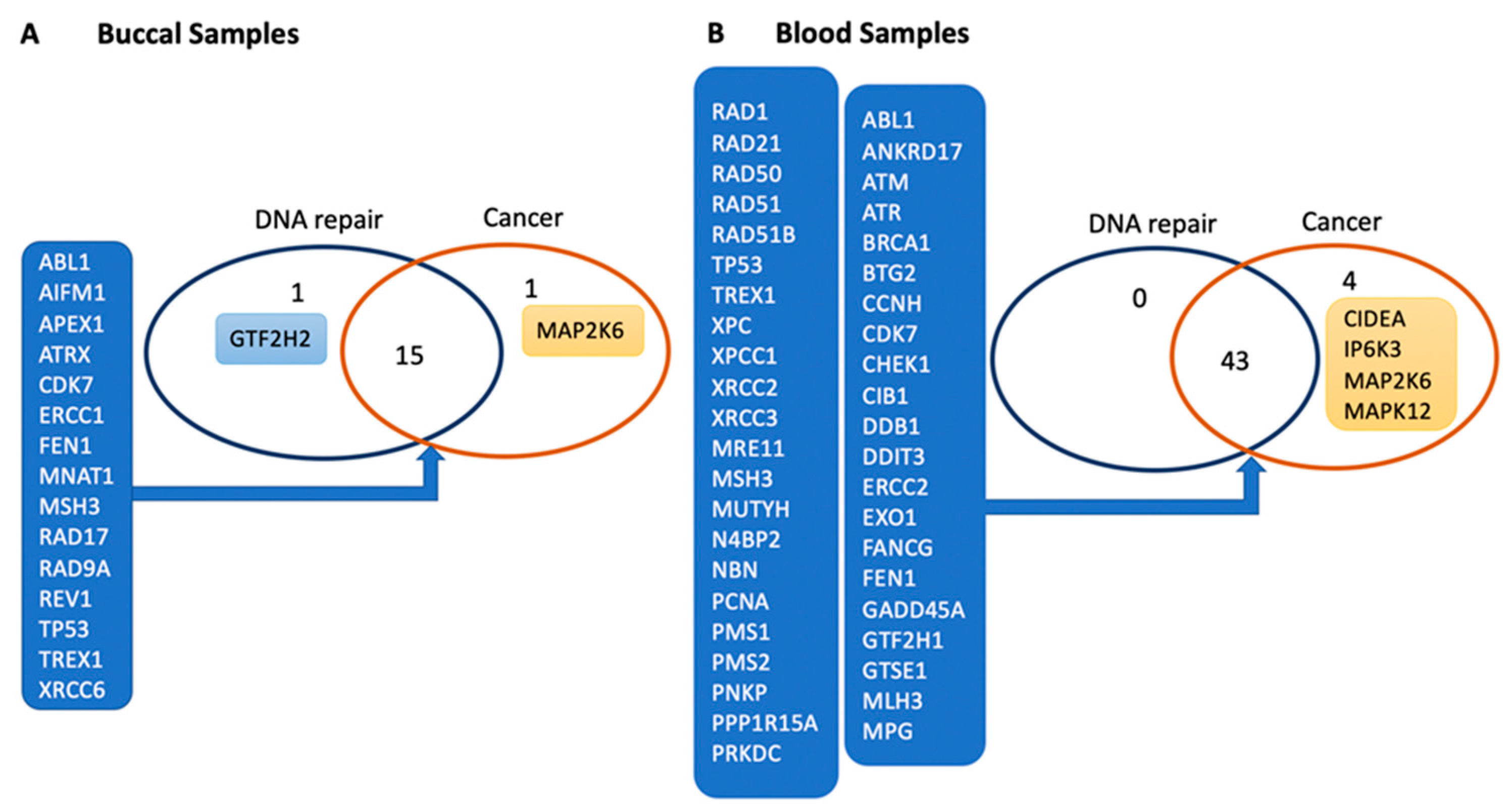
3.1. Genes Related to DNA Damage Are Differentially Regulated in Buccal and Blood Samples after E-Cig Exposure
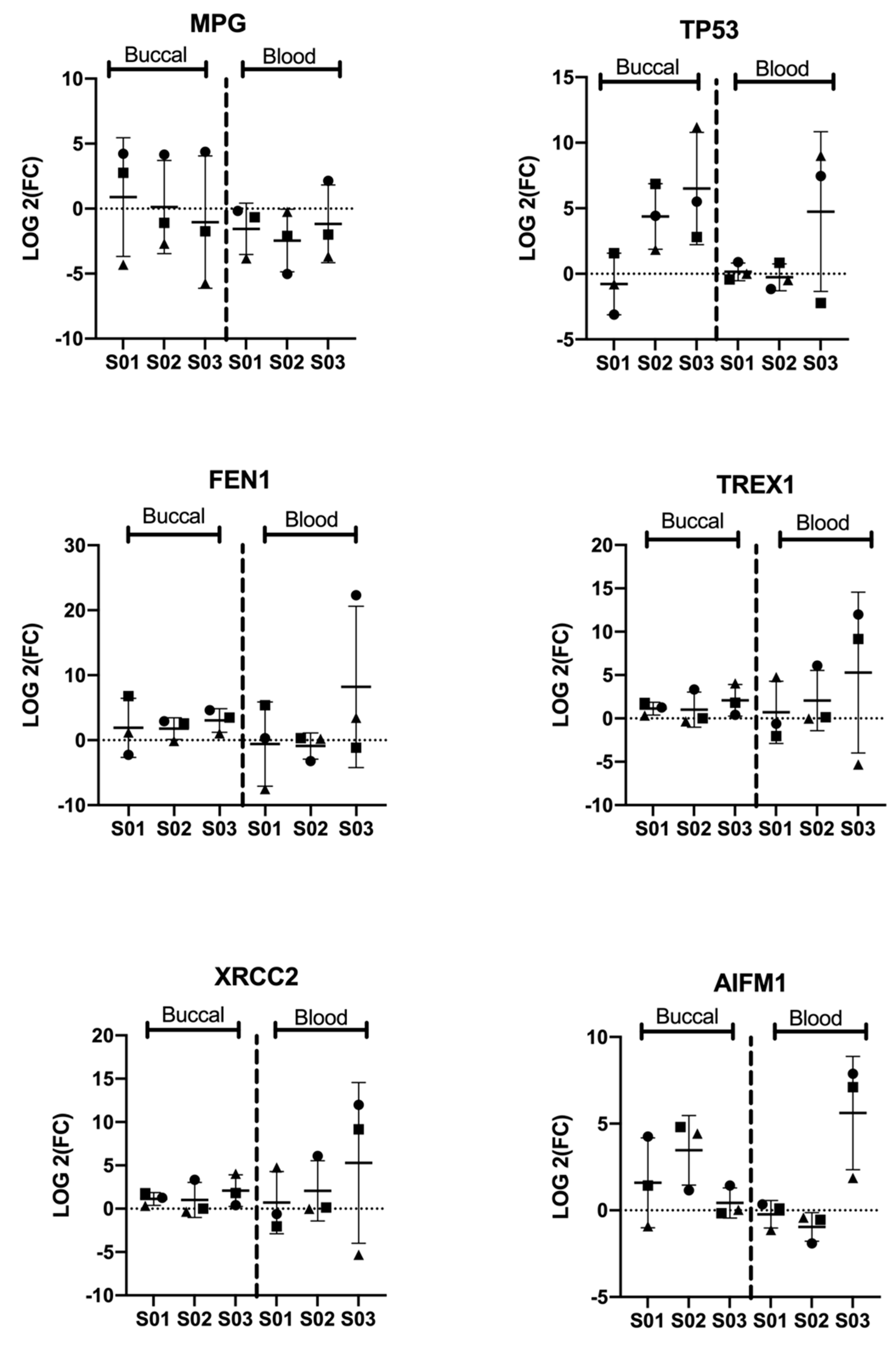
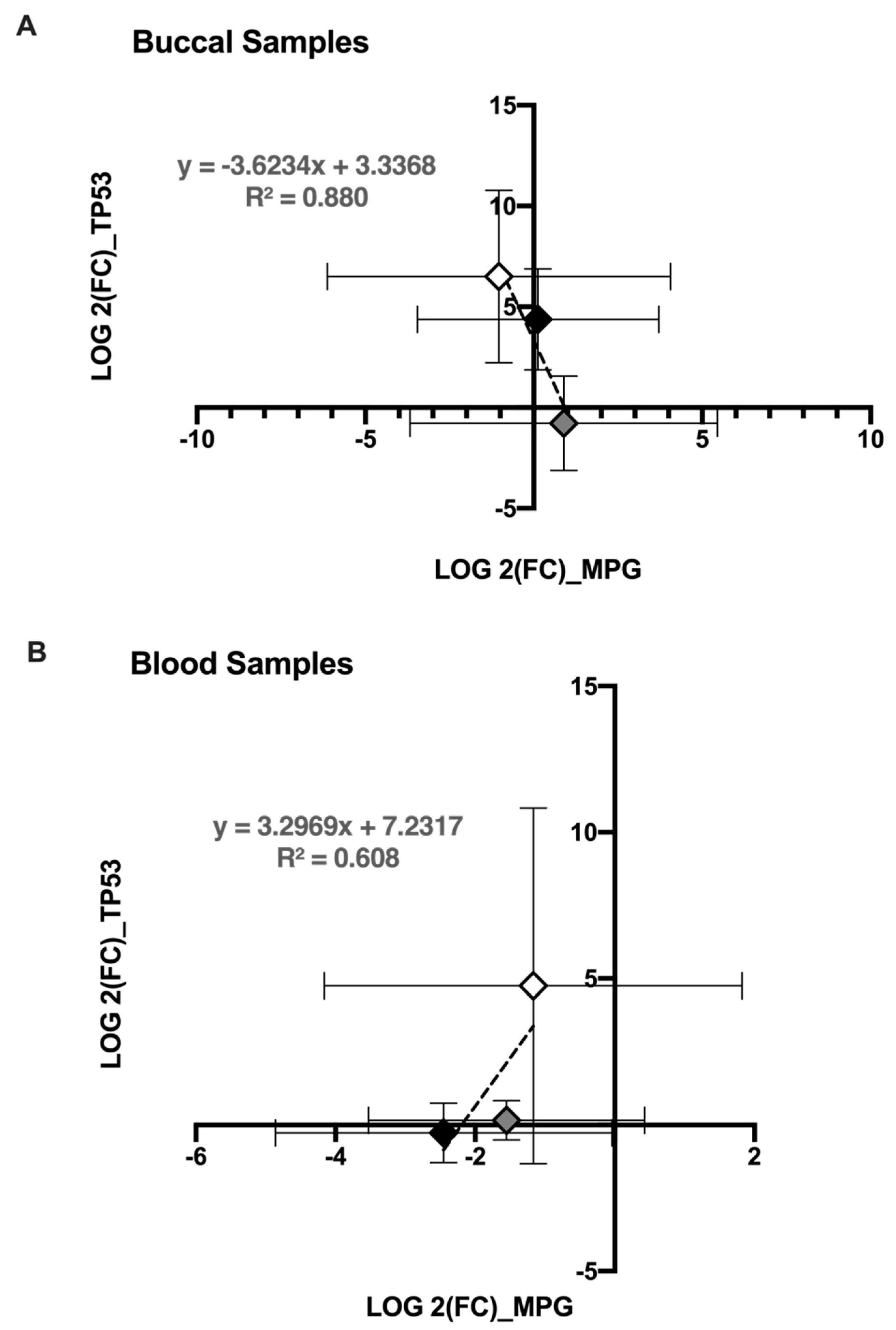
3.2. Significant Differential Expression of Genes Is Consistent among Participants
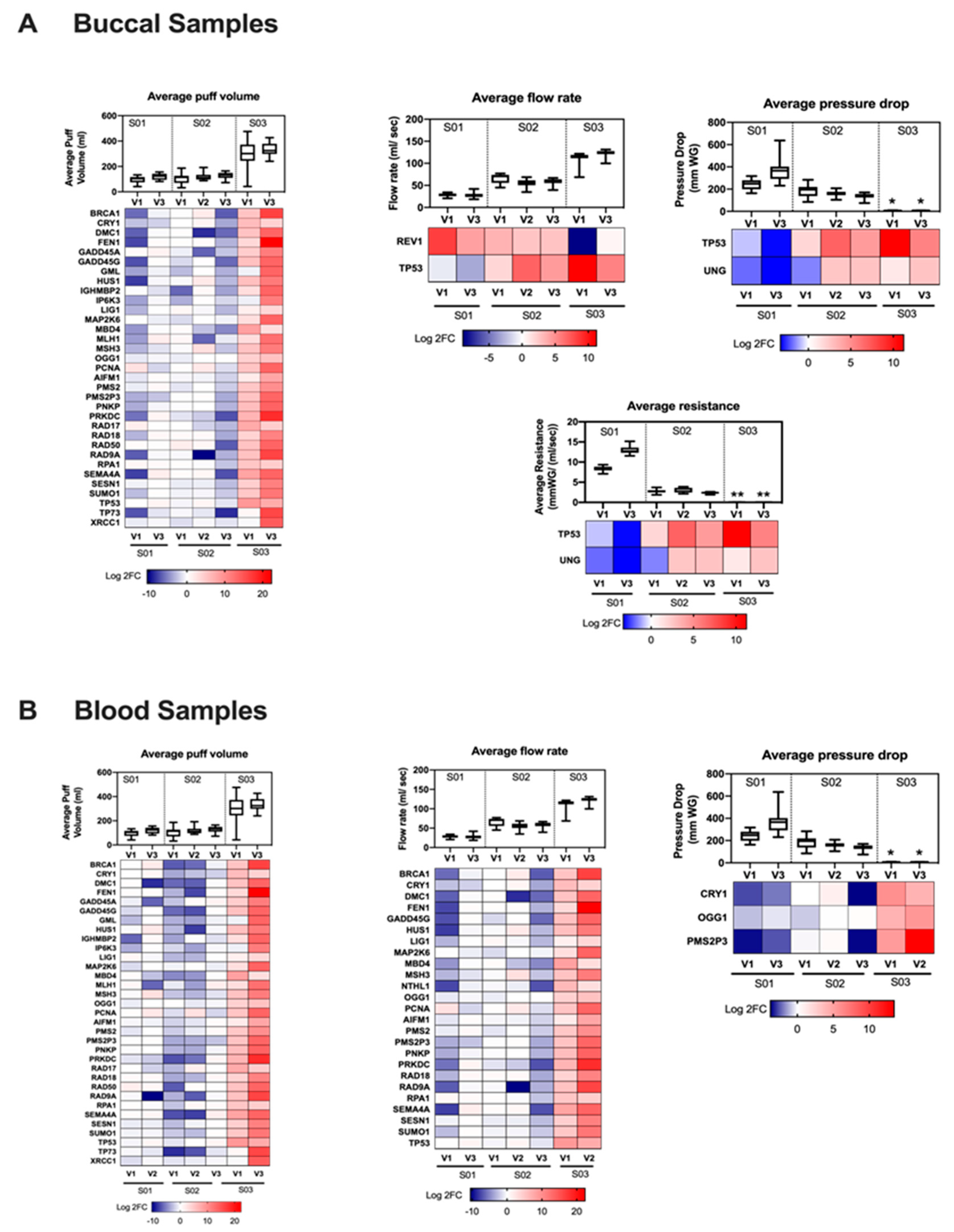
3.3. Gene Regulation Is Vaping Behavior-Dependent
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ATSDR | Agency for Toxic Substances and Disease Registry |
| BER | Base excision repair |
| CO | Carbon monoxide |
| CPT | Cell preparation tube |
| DEGs | Differentially expressed genes |
| E-cig | Electronic cigarette |
| HPHCs | Harmful and potentially harmful constituents |
| HR | Homologous recombination |
| IPA | Ingenuity pathway analysis |
| MMR | Mismatch DNA repair |
| NER | Nucleotide excision repair |
| NHEJ | Non-homologous end joining |
| PG | Propylene glycol |
| SPA-D | Smoking puff analyzer device |
| TSNAs | Tobacco specific nitrosamines |
| qPCR | Quantitative polymerase chain reaction |
| RT-PCR | Reverse transcription-polymerase chain reaction |
| VG | Vegetable glycerine |
References
- Gentzke, A.S.; Creamer, M.; Cullen, K.A.; Ambrose, B.K.; Willis, G.; Jamal, A.; King, B.A. Vital Signs: Tobacco Product Use Among Middle and High School Students—United States, 2011–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 157–164. [Google Scholar] [CrossRef]
- Adam, S. E-Cigarette Manufacturers Say New Regulations Will Devastate the Industry. Forbes. 5 May 2016. Available online: https://www.forbes.com/sites/susanadams/2016/05/05/e-cigarette-manufacturers-say-new-regulations-will-devastate-the-industry/#47d071e66d41 (accessed on 16 October 2020).
- Tegin, G.; Mekala, H.M.; Sarai, S.K.; Lippmann, S. E-Cigarette Toxicity? South Med. J. 2018, 111, 35–38. [Google Scholar] [CrossRef]
- Goniewicz, M.L.; Knysak, J.; Gawron, M.; Kosmider, L.; Sobczak, A.; Kurek, J.; Prokopowicz, A.; Jablonska-Czapla, M.; Rosik-Dulewska, C.; Havel, C.; et al. Levels of selected carcinogens and toxicants in vapour from electronic cigarettes. Tob. Control 2014, 23, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, M.; Logue, J.M.; Montesinos, V.N.; Russell, M.L.; Litter, M.I.; Gundel, L.A.; Destaillats, H. Emissions from Electronic Cigarettes: Key Parameters Affecting the Release of Harmful Chemicals. Environ. Sci. Technol. 2016, 50, 9644–9651. [Google Scholar] [CrossRef]
- Kosmider, L.; Sobczak, A.; Fik, M.; Knysak, J.; Zaciera, M.; Kurek, J.; Goniewicz, M.L. Carbonyl compounds in electronic cigarette vapors: Effects of nicotine solvent and battery output voltage. Nicotine Tob. Res. 2014, 16, 1319–1326. [Google Scholar] [CrossRef]
- Jensen, R.P.; Luo, W.; Pankow, J.F.; Strongin, R.M.; Peyton, D.H. Hidden formaldehyde in e-cigarette aerosols. N. Engl. J. Med. 2015, 372, 392–394. [Google Scholar] [CrossRef]
- Talih, S.; Balhas, Z.; Eissenberg, T.; Salman, R.; Karaoghlanian, N.; El Hellani, A.; Baalbaki, R.; Saliba, N.; Shihadeh, A. Effects of user puff topography, device voltage, and liquid nicotine concentration on electronic cigarette nicotine yield: Measurements and model predictions. Nicotine Tob. Res. 2015, 17, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Hee, S.S.Q. Formaldehyde-induced DNA adducts as biomarkers of in vitro human nasal epithelial cell exposure to formaldehyde. Mutat. Res. 2004, 563, 13–24. [Google Scholar] [CrossRef]
- Pontel, L.B.; Rosado, I.V.; Burgos-Barragan, G.; Garaycoechea, J.I.; Yu, R.; Arends, M.J.; Chandrasekaran, G.; Broecker, V.; Wei, W.; Liu, L.; et al. Endogenous Formaldehyde Is a Hematopoietic Stem Cell Genotoxin and Metabolic Carcinogen. Mol. Cell 2015, 60, 177–188. [Google Scholar] [CrossRef]
- Voulgaridou, G.-P.A.; Anestopoulos, I.; Franco, R.; Panayiotidis, M.I.; Pappa, A. DNA damage induced by endogenous aldehydes: Current state of knowledge. Mutat. Res. Mol. Mech. Mutagen 2011, 711, 13–27. [Google Scholar] [CrossRef]
- Lu, K.; Collins, L.B.; Ru, H.; Bermudez, E.; Swenberg, J.A. Distribution of DNA adducts caused by inhaled formaldehyde is consistent with induction of nasal carcinoma but not leukemia. Toxicol. Sci. 2010, 116, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Quievryn, G.; Zhitkovich, A. Loss of DNA–protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 2000, 21, 1573–1580. [Google Scholar] [CrossRef]
- Lu, K.; Ye, W.; Gold, A.; Ball, L.M.; Swenberg, J.A. Formation of S-[1-(N2-deoxyguanosinyl)methyl]glutathione between glutathione and DNA induced by formaldehyde. J. Am. Chem. Soc. 2009, 131, 3414–3415. [Google Scholar] [CrossRef]
- IARC. International Agency for Research on Cancer: Monographs on the Evaluation of Carcinogenic Risks to Humans; IARC: Lyon, France, 1988. Available online: https://www.ncbi.nlm.nih.gov/books/NBK294452/ (accessed on 16 October 2020).
- Sul, D.; Kim, H.; Oh, E.; Phark, S.; Cho, E.; Choi, S.; Kang, H.S.; Kim, E.M.; Hwang, K.W.; Jung, W.W. Gene expression profiling in lung tissues from rats exposed to formaldehyde. Arch. Toxicol. 2007, 81, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.W.; Park, S.H.; Weng, M.W.; Wang, H.T.; Huang, W.C.; Lepor, H.; Wu, X.R.; Chen, L.C.; Tang, M.S. E-cigarette smoke damages DNA and reduces repair activity in mouse lung, heart, and bladder as well as in human lung and bladder cells. Proc. Natl. Acad. Sci. USA 2018, 115, E1560–E1569. [Google Scholar] [CrossRef]
- Hecht, S.S. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat. Res. 1999, 424, 127–142. [Google Scholar] [CrossRef]
- Cloutier, J.F.; Drouin, R.; Weinfeld, M.; O’Connor, T.R.; Castonguay, A. Characterization and mapping of DNA damage induced by reactive metabolites of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) at nucleotide resolution in human genomic DNA. J. Mol. Biol. 2001, 313, 539–557. [Google Scholar] [CrossRef] [PubMed]
- Schuller, H.M. Mechanisms of smoking-related lung and pancreatic adenocarcinoma development. Nat. Rev. Cancer 2002, 2, 455–463. [Google Scholar] [CrossRef]
- ATSDR. Agency for Toxic Substances and Disease Registry, Draft Toxicological Profiles; ATSDR: Atlanta, GA, USA, 2011. Available online: http://www.atsdr.cdc.gov/toxprofiles/index.asp (accessed on 19 October 2020).
- Xue, J.; Yang, S.; Seng, S. Mechanisms of Cancer Induction by Tobacco-Specific NNK and NNN. Cancers 2014, 6, 1138–1156. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.-W.; Lee, H.-W.; Park, S.-H.; Hu, Y.; Wang, H.-T.; Chen, L.-C.; Rom, W.N.; Huang, W.C.; Lepor, H.; Wu, X.-R.; et al. Aldehydes are the predominant forces inducing DNA damage and inhibiting DNA repair in tobacco smoke carcinogenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E6152–E6161. [Google Scholar] [CrossRef]
- Prokhorova, E.A.; Egorshina, A.Y.; Zhivotovsky, B.; Kopeina, G.S. The DNA-damage response and nuclear events as regulators of nonapoptotic forms of cell death. Oncogene 2020, 39, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Loeb, L.A.; Loeb, K.R.; Anderson, J.P. Multiple mutations and cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 776–781. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Wolkowicz, M.J.; Kotova, T.; Fan, L.; Timko, M.P. Transcriptome sequencing reveals e-cigarette vapor and mainstream-smoke from tobacco cigarettes activate different gene expression profiles in human bronchial epithelial cells. Sci. Rep. 2016, 6, 23984. [Google Scholar] [CrossRef][Green Version]
- Martin, E.M.; Clapp, P.W.; Rebuli, M.E.; Pawlak, E.A.; Glista-Baker, E.; Benowitz, N.L.; Fry, R.C.; Jaspers, I. E-cigarette use results in suppression of immune and inflammatory-response genes in nasal epithelial cells similar to cigarette smoke. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 311, L135–L144. [Google Scholar] [CrossRef] [PubMed]
- Herr, C.; Tsitouras, K.; Niederstraßer, J.; Backes, C.; Beisswenger, C.; Dong, L.; Guillot, L.; Keller, A.; Bals, R. Cigarette smoke and electronic cigarettes differentially activate bronchial epithelial cells. Respir. Res. 2020, 21, 67. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagen 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed]
- Vlahopoulos, S.; Adamaki, M.; Khoury, N.; Zoumpourlis, V.; Boldogh, I. Roles of DNA repair enzyme OGG1 in innate immunity and its significance for lung cancer. Pharmacol. Ther. 2019, 194, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb. Perspect. Med. 2016, 6. [Google Scholar] [CrossRef]
- Kim, I.S.; Lee, M.Y.; Lee, I.H.; Shin, S.L.; Lee, S.Y. Gene expression of flap endonuclease-1 during cell proliferation and differentiation. Biochim. Biophys. Acta 2000, 1496, 333–340. [Google Scholar] [CrossRef][Green Version]
- Faridounnia, M.; Folkers, G.E.; Boelens, R. Function and Interactions of ERCC1-XPF in DNA Damage Response. Molecules 2018, 23, 3205. [Google Scholar] [CrossRef]
- Vogelstein, B.; Lane, D.; Levine, A.J. Surfing the p53 network. Nature 2000, 408, 307–310. [Google Scholar] [CrossRef]
- Pietenpol, J.A.; Tokino, T.; Thiagalingam, S.; El-Deiry, W.S.; Kinzler, K.W.; Vogelstein, B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc. Natl. Acad. Sci. USA 1994, 91, 1998–2002. [Google Scholar] [CrossRef]
- Resnick-Silverman, L.; St Clair, S.; Maurer, M.; Zhao, K.; Manfredi, J.J. Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev. 1998, 12, 2102–2107. [Google Scholar] [CrossRef] [PubMed]
- Fishel, M.L.; He, Y.; Smith, M.L.; Kelley, M.R. Manipulation of base excision repair to sensitize ovarian cancer cells to alkylating agent temozolomide. Clin. Cancer Res. 2007, 13, 260–267. [Google Scholar] [CrossRef]
- Harrison, J.F.; Rinne, M.L.; Kelley, M.R.; Druzhyna, N.M.; Wilson, G.L.; Ledoux, S.P. Altering DNA base excision repair: Use of nuclear and mitochondrial-targeted N-methylpurine DNA glycosylase to sensitize astroglia to chemotherapeutic agents. Glia 2007, 55, 1416–1425. [Google Scholar] [CrossRef] [PubMed]
- Cerda, S.R.; Turk, P.W.; Thor, A.D.; Weitzman, S.A. Altered expression of the DNA repair protein, N-methylpurine-DNA glycosylase (MPG) in breast cancer. FEBS Lett. 1998, 431, 12–18. [Google Scholar] [CrossRef]
- Rinne, M.; Caldwell, D.; Kelley, M.R. Transient adenoviral N-methylpurine DNA glycosylase overexpression imparts chemotherapeutic sensitivity to human breast cancer cells. Mol. Cancer Ther. 2004, 3, 955–967. [Google Scholar] [PubMed]
- Liu, C.; Tu, Y.; Yuan, J.; Mao, X.; He, S.; Wang, L.; Fu, G.; Zong, J.; Zhang, Y. Aberrant expression of N-methylpurine-DNA glycosylase influences patient survival in malignant gliomas. J. Biomed. Biotechnol. 2012, 2012, 760679. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Xing, G.; Yuan, L.; Wang, J.; Wang, S.; Yin, Y.; Tian, C.; He, F.; Zhang, L. N-methylpurine DNA glycosylase inhibits p53-mediated cell cycle arrest and coordinates with p53 to determine sensitivity to alkylating agents. Cell Res. 2012, 22, 1285–1303. [Google Scholar] [CrossRef][Green Version]
- Bridges, R.B.; Humble, J.W.; Turbek, J.A.; Rehm, S.R. Smoking history, cigarette yield and smoking behavior as determinants of smoke exposure. Eur. J. Respir. Dis. Suppl. 1986, 146, 129–137. [Google Scholar] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamad, S.H.; Brinkman, M.C.; Tsai, Y.-H.; Mellouk, N.; Cross, K.; Jaspers, I.; Clark, P.I.; Granville, C.A. Pilot Study to Detect Genes Involved in DNA Damage and Cancer in Humans: Potential Biomarkers of Exposure to E-Cigarette Aerosols. Genes 2021, 12, 448. https://doi.org/10.3390/genes12030448
Hamad SH, Brinkman MC, Tsai Y-H, Mellouk N, Cross K, Jaspers I, Clark PI, Granville CA. Pilot Study to Detect Genes Involved in DNA Damage and Cancer in Humans: Potential Biomarkers of Exposure to E-Cigarette Aerosols. Genes. 2021; 12(3):448. https://doi.org/10.3390/genes12030448
Chicago/Turabian StyleHamad, Samera H., Marielle C. Brinkman, Yi-Hsuan Tsai, Namya Mellouk, Kandice Cross, Ilona Jaspers, Pamela I. Clark, and Courtney A. Granville. 2021. "Pilot Study to Detect Genes Involved in DNA Damage and Cancer in Humans: Potential Biomarkers of Exposure to E-Cigarette Aerosols" Genes 12, no. 3: 448. https://doi.org/10.3390/genes12030448
APA StyleHamad, S. H., Brinkman, M. C., Tsai, Y.-H., Mellouk, N., Cross, K., Jaspers, I., Clark, P. I., & Granville, C. A. (2021). Pilot Study to Detect Genes Involved in DNA Damage and Cancer in Humans: Potential Biomarkers of Exposure to E-Cigarette Aerosols. Genes, 12(3), 448. https://doi.org/10.3390/genes12030448