
Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis


{kind=link}
Abstract
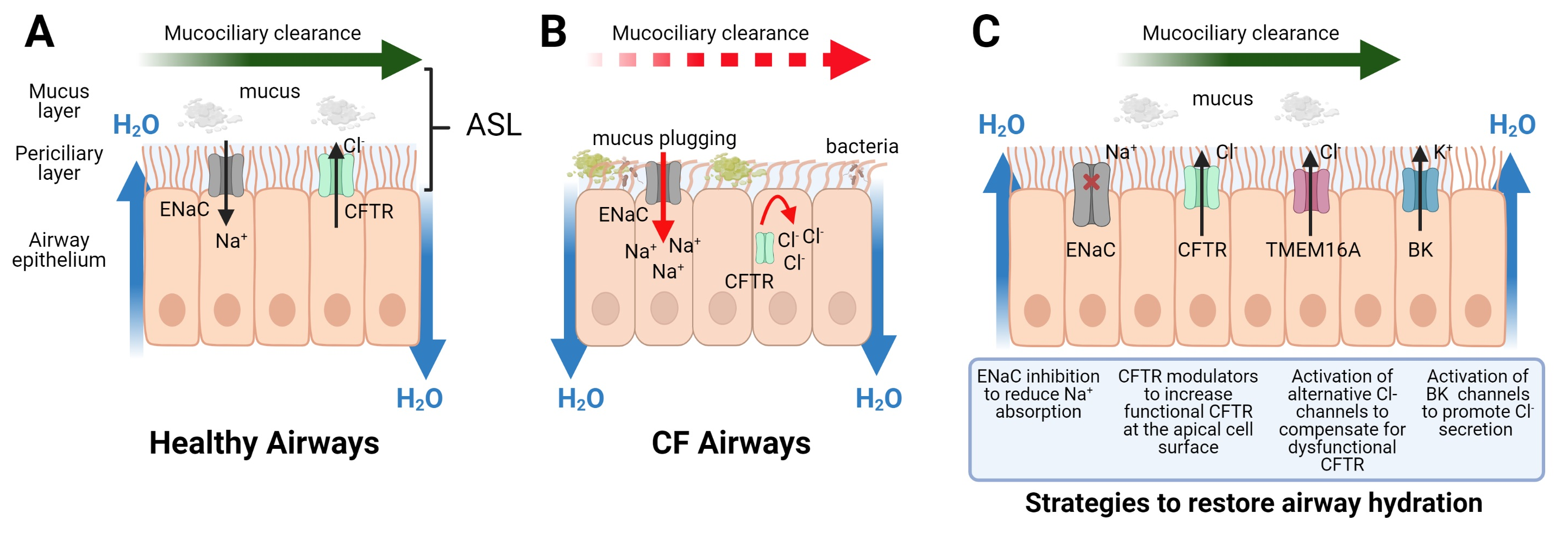
:1. Introduction
2. Ion Channel Targets
2.1. CFTR Modulators
2.2. Approved CFTR Modulator Therapies—Current Challenges
3. TMEM16A
4. ENaC
4.1. Direct ENaC Blockers
4.2. Protease Inhibitors
4.3. Reduction of Surface ENaC Expression
4.4. Additional Approaches to Target ENaC
5. BK Channels
6. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Webster, M.J.; Tarran, R. Slippery When Wet: Airway Surface Liquid Homeostasis and Mucus Hydration. Curr. Top. Membr. 2018, 81, 293–335. [Google Scholar] [CrossRef]
- Boucher, R.C. Evidence for airway surface dehydration as the initiating event in CF airway disease. J. Intern. Med. 2007, 261, 5–16. [Google Scholar] [CrossRef]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.; Burton, B.; Cao, D.; Neuberger, T.; Turnbull, A.; Singh, A.; Joubran, J.; Hazlewood, A.; et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc. Natl. Acad. Sci. USA 2009, 106, 18825–18830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramsey, B.W.; Davies, J.; McElvaney, N.G.; Tullis, E.; Bell, S.C.; Drevinek, P.; Griese, M.; McKone, E.F.; Wainwright, C.E.; Konstan, M.W.; et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N. Engl. J. Med. 2011, 365, 1663–1672. [Google Scholar] [CrossRef] [Green Version]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordonez, C.L.; Geller, D.E.; Group, V.X.S. Ivacaftor in subjects with cystic fibrosis who are homozygous for the F508del-CFTR mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R., Jr.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator ivacaftor abrogates pharmacological correction of DeltaF508 CFTR in cystic fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veit, G.; Avramescu, R.G.; Perdomo, D.; Phuan, P.W.; Bagdany, M.; Apaja, P.M.; Borot, F.; Szollosi, D.; Wu, Y.S.; Finkbeiner, W.E.; et al. Some gating potentiators, including VX-770, diminish DeltaF508-CFTR functional expression. Sci. Transl. Med. 2014, 6, 246ra97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W. Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 1783–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graeber, S.Y.; Dopfer, C.; Naehrlich, L.; Gyulumyan, L.; Scheuermann, H.; Hirtz, S.; Wege, S.; Mairbaurl, H.; Dorda, M.; Hyde, R.; et al. Effects of Lumacaftor-Ivacaftor Therapy on Cystic Fibrosis Transmembrane Conductance Regulator Function in Phe508del Homozygous Patients with Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2018, 197, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and safety of the elexacaftor plus tezacaftor plus ivacaftor combination regimen in people with cystic fibrosis homozygous for the F508del mutation: A double-blind, randomised, phase 3 trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Drevinek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Solomon, G.M.; Zeitlin, P.L.; Flume, P.A.; Casey, A.; McCoy, K.; Zemanick, E.T.; Mandagere, A.; Troha, J.M.; Shoemaker, S.A.; et al. Pharmacokinetics and safety of cavosonstat (N91115) in healthy and cystic fibrosis adults homozygous for F508DEL-CFTR. J. Cyst. Fibros. 2017, 16, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Giuliano, K.A.; Wachi, S.; Drew, L.; Dukovski, D.; Green, O.; Bastos, C.; Cullen, M.D.; Hauck, S.; Tait, B.D.; Munoz, B.; et al. Use of a High-Throughput Phenotypic Screening Strategy to Identify Amplifiers, a Novel Pharmacological Class of Small Molecules That Exhibit Functional Synergy with Potentiators and Correctors. SLAS Discov. 2018, 23, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Molinski, S.V.; Ahmadi, S.; Ip, W.; Ouyang, H.; Villella, A.; Miller, J.P.; Lee, P.S.; Kulleperuma, K.; Du, K.; Di Paola, M.; et al. Orkambi® and amplifier co-therapy improves function from a rare CFTR mutation in gene-edited cells and patient tissue. EMBO Mol. Med. 2017, 9, 1224–1243. [Google Scholar] [CrossRef]
- Xue, X.; Mutyam, V.; Tang, L.; Biswas, S.; Du, M.; Jackson, L.A.; Dai, Y.; Belakhov, V.; Shalev, M.; Chen, F.; et al. Synthetic aminoglycosides efficiently suppress cystic fibrosis transmembrane conductance regulator nonsense mutations and are enhanced by ivacaftor. Am. J. Respir. Cell Mol. Biol. 2014, 50, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Crawford, D.K.; Mullenders, J.; Pott, J.; Boj, S.F.; Landskroner-Eiger, S.; Goddeeris, M.M. Targeting G542X CFTR nonsense alleles with ELX-02 restores CFTR function in human-derived intestinal organoids. J. Cyst. Fibros. 2021. [Google Scholar] [CrossRef]
- Hisert, K.B.; Heltshe, S.L.; Pope, C.; Jorth, P.; Wu, X.; Edwards, R.M.; Radey, M.; Accurso, F.J.; Wolter, D.J.; Cooke, G.; et al. Restoring Cystic Fibrosis Transmembrane Conductance Regulator Function Reduces Airway Bacteria and Inflammation in People with Cystic Fibrosis and Chronic Lung Infections. Am. J. Respir. Crit. Care Med. 2017, 195, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Strug, L.J.; Gonska, T.; He, G.; Keenan, K.; Ip, W.; Boelle, P.Y.; Lin, F.; Panjwani, N.; Gong, J.; Li, W.; et al. Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum. Mol. Genet. 2016, 25, 4590–4600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trinh, N.T.; Bilodeau, C.; Maille, E.; Ruffin, M.; Quintal, M.C.; Desrosiers, M.Y.; Rousseau, S.; Brochiero, E. Deleterious impact of Pseudomonas aeruginosa on cystic fibrosis transmembrane conductance regulator function and rescue in airway epithelial cells. Eur. Respir. J. 2015, 45, 1590–1602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maille, E.; Ruffin, M.; Adam, D.; Messaoud, H.; Lafayette, S.L.; McKay, G.; Nguyen, D.; Brochiero, E. Quorum Sensing Down-Regulation Counteracts the Negative Impact of Pseudomonas aeruginosa on CFTR Channel Expression, Function and Rescue in Human Airway Epithelial Cells. Front. Cell Infect. Microbiol. 2017, 7, 470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Knowles, M.R.; Clarke, L.L.; Boucher, R.C. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N. Engl. J. Med. 1991, 325, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Ratjen, F.; Durham, T.; Navratil, T.; Schaberg, A.; Accurso, F.J.; Wainwright, C.; Barnes, M.; Moss, R.B.; Group, T.-S.I. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 539–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Button, B.; Okada, S.F.; Frederick, C.B.; Thelin, W.R.; Boucher, R.C. Mechanosensitive ATP release maintains proper mucus hydration of airways. Sci. Signal. 2013, 6, ra46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A Potentiation: A Novel Therapeutic Approach for the Treatment of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 946–954. [Google Scholar] [CrossRef] [Green Version]
- Kunzelmann, K.; Ousingsawat, J.; Cabrita, I.; Dousova, T.; Bahr, A.; Janda, M.; Schreiber, R.; Benedetto, R. TMEM16A in Cystic Fibrosis: Activating or Inhibiting? Front. Pharmacol. 2019, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Cabrita, I.; Benedetto, R.; Wanitchakool, P.; Lerias, J.; Centeio, R.; Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. TMEM16A Mediates Mucus Production in Human Airway Epithelial Cells. Am. J. Respir. Cell Mol. Biol. 2021, 64, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [Green Version]
- Danahay, H.; Fox, R.; Lilley, S.; Charlton, H.; Adley, K.; Christie, L.; Ansari, E.; Ehre, C.; Flen, A.; Tuvim, M.J.; et al. Potentiating TMEM16A does not stimulate airway mucus secretion or bronchial and pulmonary arterial smooth muscle contraction. FASEB Bioadv. 2020, 2, 464–477. [Google Scholar] [CrossRef]
- Boucher, R.C.; Stutts, M.J.; Knowles, M.R.; Cantley, L.; Gatzy, J.T. Na+ transport in cystic fibrosis respiratory epithelia. Abnormal basal rate and response to adenylate cyclase activation. J. Clin. Investig. 1986, 78, 1245–1252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mall, M.; Grubb, B.R.; Harkema, J.R.; O'Neal, W.K.; Boucher, R.C. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat. Med. 2004, 10, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Kleyman, T.R.; Carattino, M.D.; Hughey, R.P. ENaC at the cutting edge: Regulation of epithelial sodium channels by proteases. J. Biol. Chem. 2009, 284, 20447–20451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reihill, J.A.; Walker, B.; Hamilton, R.A.; Ferguson, T.E.; Elborn, J.S.; Stutts, M.J.; Harvey, B.J.; Saint-Criq, V.; Hendrick, S.M.; Martin, S.L. Inhibition of Protease-Epithelial Sodium Channel Signaling Improves Mucociliary Function in Cystic Fibrosis Airways. Am. J. Respir. Crit. Care Med. 2016, 194, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Myerburg, M.M.; McKenna, E.E.; Luke, C.J.; Frizzell, R.A.; Kleyman, T.R.; Pilewski, J.M. Prostasin expression is regulated by airway surface liquid volume and is increased in cystic fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 294, L932–L941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentzsch, M.; Dang, H.; Dang, Y.; Garcia-Caballero, A.; Suchindran, H.; Boucher, R.C.; Stutts, M.J. The cystic fibrosis transmembrane conductance regulator impedes proteolytic stimulation of the epithelial Na+ channel. J. Biol. Chem. 2010, 285, 32227–32232. [Google Scholar] [CrossRef] [Green Version]
- Burrows, E.; Southern, K.W.; Noone, P. Sodium channel blockers for cystic fibrosis. Cochrane Database Syst. Rev. 2006, CD005087. [Google Scholar] [CrossRef] [Green Version]
- Nickolaus, P.; Jung, B.; Sabater, J.; Constant, S.; Gupta, A. Preclinical evaluation of the epithelial sodium channel inhibitor BI 1265162 for treatment of cystic fibrosis. ERJ Open Res. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Coote, K.; Atherton-Watson, H.C.; Sugar, R.; Young, A.; MacKenzie-Beevor, A.; Gosling, M.; Bhalay, G.; Bloomfield, G.; Dunstan, A.; Bridges, R.J.; et al. Camostat attenuates airway epithelial sodium channel function in vivo through the inhibition of a channel-activating protease. J. Pharmacol. Exp. Ther. 2009, 329, 764–774. [Google Scholar] [CrossRef] [Green Version]
- Rowe, S.M.; Reeves, G.; Hathorne, H.; Solomon, G.M.; Abbi, S.; Renard, D.; Lock, R.; Zhou, P.; Danahay, H.; Clancy, J.P.; et al. Reduced sodium transport with nasal administration of the prostasin inhibitor camostat in subjects with cystic fibrosis. Chest 2013, 144, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Caballero, A.; Rasmussen, J.E.; Gaillard, E.; Watson, M.J.; Olsen, J.C.; Donaldson, S.H.; Stutts, M.J.; Tarran, R. SPLUNC1 regulates airway surface liquid volume by protecting ENaC from proteolytic cleavage. Proc. Natl. Acad. Sci. USA 2009, 106, 11412–11417. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, C.A.; Blanchard, M.G.; Alijevic, O.; Tan, C.D.; Kellenberger, S.; Bencharit, S.; Cao, R.; Kesimer, M.; Walton, W.G.; Henderson, A.G.; et al. Identification of the SPLUNC1 ENaC-inhibitory domain yields novel strategies to treat sodium hyperabsorption in cystic fibrosis airway epithelial cultures. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 305, L990–L1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.W.; Walker, M.P.; Sesma, J.; Wu, B.; Stuhlmiller, T.J.; Sabater, J.R.; Abraham, W.M.; Crowder, T.M.; Christensen, D.J.; Tarran, R. SPX-101 Is a Novel Epithelial Sodium Channel-targeted Therapeutic for Cystic Fibrosis That Restores Mucus Transport. Am. J. Respir. Crit. Care Med. 2017, 196, 734–744. [Google Scholar] [CrossRef] [PubMed]
- Couroux, P.; Farias, P.; Rizvi, L.; Griffin, K.; Hudson, C.; Crowder, T.; Tarran, R.; Tullis, E. First clinical trials of novel ENaC targeting therapy, SPX-101, in healthy volunteers and adults with cystic fibrosis. Pulm. Pharmacol. Ther. 2019, 58, 101819. [Google Scholar] [CrossRef] [PubMed]
- Crosby, J.R.; Zhao, C.; Jiang, C.; Bai, D.; Katz, M.; Greenlee, S.; Kawabe, H.; McCaleb, M.; Rotin, D.; Guo, S.; et al. Inhaled ENaC antisense oligonucleotide ameliorates cystic fibrosis-like lung disease in mice. J. Cyst. Fibros. 2017, 16, 671–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manzanares, D.; Gonzalez, C.; Ivonnet, P.; Chen, R.S.; Valencia-Gattas, M.; Conner, G.E.; Larsson, H.P.; Salathe, M. Functional apical large conductance, Ca2+-activated, and voltage-dependent K+ channels are required for maintenance of airway surface liquid volume. J. Biol. Chem. 2011, 286, 19830–19839. [Google Scholar] [CrossRef] [Green Version]
- Manzanares, D.; Krick, S.; Baumlin, N.; Dennis, J.S.; Tyrrell, J.; Tarran, R.; Salathe, M. Airway Surface Dehydration by Transforming Growth Factor beta (TGF-beta) in Cystic Fibrosis Is Due to Decreased Function of a Voltage-dependent Potassium Channel and Can Be Rescued by the Drug Pirfenidone. J. Biol. Chem. 2015, 290, 25710–25716. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.D.; Baumlin, N.; Yoshida, M.; Polineni, D.; Salathe, S.F.; David, J.K.; Peloquin, C.A.; Wanner, A.; Dennis, J.S.; Sailland, J.; et al. Losartan Rescues Inflammation-related Mucociliary Dysfunction in Relevant Models of Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 313–324. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reihill, J.A.; Douglas, L.E.J.; Martin, S.L. Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis. Genes 2021, 12, 453. https://doi.org/10.3390/genes12030453
Reihill JA, Douglas LEJ, Martin SL. Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis. Genes. 2021; 12(3):453. https://doi.org/10.3390/genes12030453
Chicago/Turabian StyleReihill, James A., Lisa E. J. Douglas, and S. Lorraine Martin. 2021. "Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis" Genes 12, no. 3: 453. https://doi.org/10.3390/genes12030453
APA StyleReihill, J. A., Douglas, L. E. J., & Martin, S. L. (2021). Modulation of Ion Transport to Restore Airway Hydration in Cystic Fibrosis. Genes, 12(3), 453. https://doi.org/10.3390/genes12030453