
Heterogeneity of the Clinical Presentation of the MEN1 LRG_509 c.781C>T (p.Leu261Phe) Variant Within a Three-Generation Family

 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
3. Results
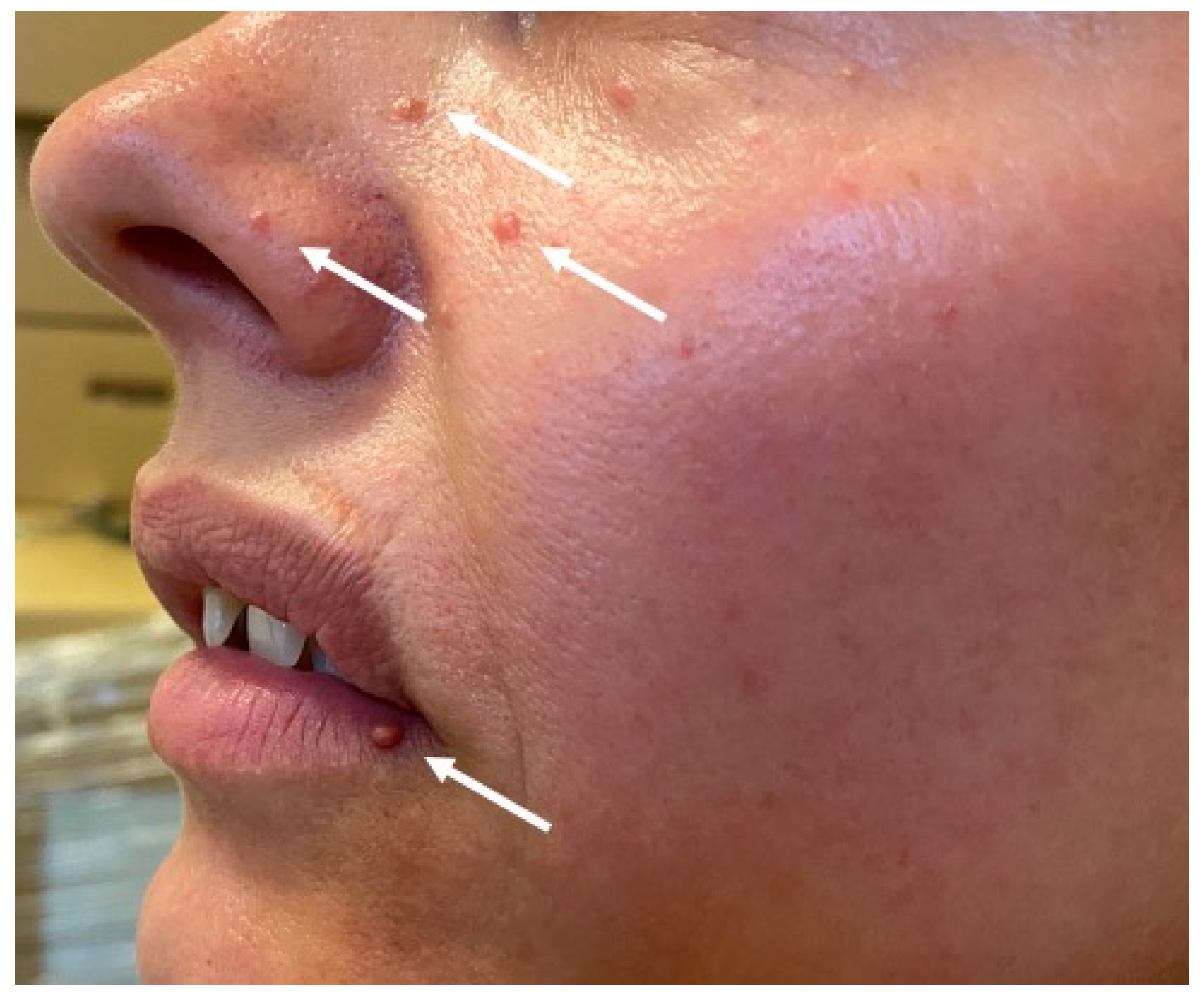
3.1. Index Patient
3.2. Patient II.4
3.3. Patient III.3
3.4. Patient III.5
3.5. Patient III.9
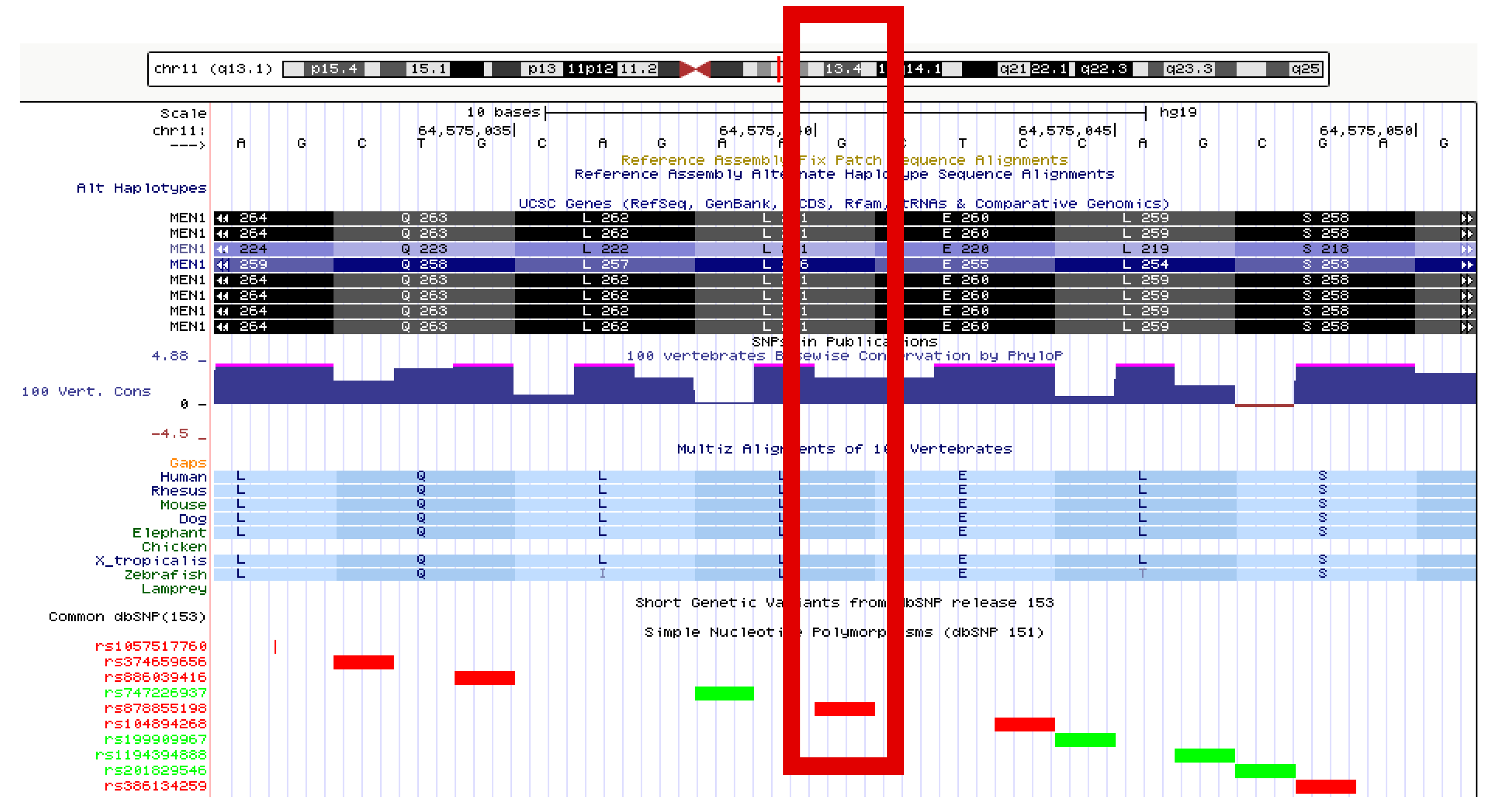
3.6. Genetic Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol. Cell Endocrinol. 2014, 386, 2–15. [Google Scholar] [CrossRef] [PubMed]
- Romanet, P.; Philibert, P.; Fina, F.; Cuny, T.; Roche, C.; Ouafik, L.; Paris, F.; Reynaud, R.; Barlier, A. Using Digital Droplet Polymerase Chain Reaction to Detect the Mosaic GNAS Mutations in Whole Blood DNA or Circulating Cell-Free DNA in Fibrous Dysplasia and McCune-Albright Syndrome. J. Pediatr. 2019, 205, 281–285.e4. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.M.; Zhang, Q.; Huang, L.H.; Mo, Z.H.; Long, X.D.; Yang, Y.B.; Yang, W.J.; Liu, J.; Jin, P. Identification of Novel Variants in MEN1: A Study Conducted with Four Multiple Endocrine Neoplasia Type 1 Patients. Horm. Metab. Res. 2020. [Google Scholar] [CrossRef]
- Concolino, P.; Costella, A.; Capoluongo, E. Multiple endocrine neoplasia type 1 (MEN1): An update of 208 new germline variants reported in the last nine years. Cancer Genet. 2016, 209, 36–41. [Google Scholar] [CrossRef]
- Lemos, M.C.; Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1): Analysis of 1336 mutations reported in the first decade following identification of the gene. Hum. Mutat. 2018, 29, 22–32. [Google Scholar] [CrossRef]
- Rogoziński, D.; Gilis-Januszewska, A.; Skalniak, A.; Kluczyński, Ł.; Pantofliński, J.; Hubalewska-Dydejczyk, A. Pituitary tumors in MEN1 syndrome—The new insight into the diagnosis and treatment. Endokrynol. Pol. 2019, 70, 445–452. [Google Scholar] [CrossRef]
- Soczomski, P.W.; Jurecka-Lubieniecka, B.; Rogozik, N.; Tukiendorf, A.; Jarząb, B.; Bednarczuk, T. Multiple Endocrine Neoplasia type 1 in Poland: A two center experience. Endokrynol. Pol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Godlewska, M.; Boguslawska, A.; Nowak, A.; Skalniak, A.; Sowa-Staszczak, A.; Gilis-Januszewska, A.; Hubalewska-Dydejczyk, A. Acromegaly and late-onset primary hyperparathyroidism in a female with a rare MEN1 gene variant of yet undetermined clinical significance (p.Val167Ala). Endokrynol. Pol. 2020, 71, 579–580. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V. Multiple endocrine neoplasia type 1. Indian J. Endocrinol. Metab. 2012, 16, 272. [Google Scholar]
- Mauchlen, R.; Carty, D.; Talla, M.; Drummond, R. Multiple endocrine neoplasia type 1 (MEN1) mosaicism caused by a c.124G>A variant in the MEN1 gene. Endocr. Abstr. 2019. [Google Scholar] [CrossRef]
- Beijers, H.J.B.H.; Stikkelbroeck, N.M.L.; Mensenkamp, A.R.; Pfundt, R.; van der Luijt, R.B.; Timmers, H.J.L.M.; Hermus, A.R.M.M.; Kempers, M.J.E. Germline and somatic mosaicism in a family with multiple endocrine neoplasia type 1 (MEN1) syndrome. Eur. J. Endocrinol. 2019, 180, K15–K19. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; Melmed, S.; Sakurai, A.; Tonelli, F.; Brandi, M.L. MEN1 2012 Guideline. J. Clin. Endocrinol. Metab. 2012. [Google Scholar] [CrossRef]
- Cavaco, B.M.; Domingues, R.; Bacelar, M.C.; Cardoso, H.; Barros, L.; Gomes, L.; Ruas, M.M.A.; Agapito, A.; Garrao, A.; Pannet, A.A.J. Mutational analysis of Portuguese families with multiple endocrine neoplasia type 1 reveals large germline deletions. Clin. Endocrinol. (Oxf.) 2002, 56, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V. Multiple endocrine neoplasia type 1 (MEN1). Best Pract. Res. Clin. Endocrinol. Metab. 2010, 24, 355–370. [Google Scholar] [CrossRef]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Lemmens, I.; van de ven Wim, J.M.; Kas, K.; Zhabg, C.X.; Giraud, S.; Waotot, V.; Buisson, N.; de Witte, D.; Salandre, J.; Lenoir, G. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. Hum. Mol. Genet. 1997, 6, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Tham, E.; Grandell, U.; Lindgren, E.; Toss, G.; Skogseid, B.; Nordenskjöld, M. Clinical testing for mutations in the MEN1 gene in Sweden: A report on 200 unrelated cases. J. Clin. Endocrinol. Metab. 2007, 92, 3389–3395. [Google Scholar] [CrossRef] [Green Version]
- Concolino, P.; Rossodivita, A.; Carrozza, C.; Rafaelli, M.; Lombardi, C.P.; Rigante, D.; Pitocco, D.; Stabile, A.; Bellantone, R.; Zuppi, C. A novel MEN1 frameshift germline mutation in two Italian monozygotic twins. Clin. Chem. Lab. Med. 2008, 46, 824–826. [Google Scholar] [CrossRef]
- Flanagan, D.E.H.; Armitage, M.; Clein, G.P.; Thakker, R.V. Prolactinoma presenting in identical twins with multiple endocrine neoplasia type 1. Clin. Endocrinol. (Oxf.) 1996, 45, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Vergès, B.; Boureille, F.; Goudet, P.; Murat, A.; Beckers, A.; Sassolas, G.; Cougard, P.; Chambe, B.; Montvernay, C.; Calender, A. Pituitary Disease in MEN Type 1 (MEN1): Data from the France-Belgium MEN1 Multicenter Study. J. Clin. Endocrinol. Metab. 2002, 87, 457–465. [Google Scholar] [CrossRef]
- Trouillas, J.; Labat-Moleur, F.; Sturm, N.; Kujas, M.; Heymann, M.F.; Figarella-Branger, D.; Patey, M.; Mazucca, M.; Decullier, E. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): A case-control study in a series of 77 patients versus 2509 non-MEN1 patients. Am. J. Surg. Pathol. 2008, 32, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Goudet, P.; Dalac, A.; Le Bras, M.; Cardot-Bauters, C.; Nicolli, P.; Levy-Bohbot, N.; du Boullay, H.; Bertagna, X.; Ruszniewski, P. MEN1 disease occurring before 21 years old: A 160-patient cohort study from the Groupe d’étude des Tumeurs Endocrines. J. Clin. Endocrinol. Metab. 2015, 100, 1568–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lourenço, D.M.; Toledo, R.A.; Mackowiak, I.I.; Coutinho, F.L.; Cavalcanti, M.G.; Correia-Deur, J.E.M.; Montenegro, F.; Siqueira, S.A.C.; Margarido, L.C.; Machado, M.C. Multiple endocrine neoplasia type 1 in Brazil: MEN1 founding mutation, clinical features, and bone mineral density profile. Eur. J. Endocrinol. 2018, 159, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, R.; Green, J.; Marx, S.J. Primary hyperparathyroidism in familial multiple endocrine neoplasia type I. Long-term follow-up of serum calcium levels after parathyroidectomy. Am. J. Med. 1985, 78, 467–474. [Google Scholar] [CrossRef]
- Marx, S.J.; Simonds, W.F. Hereditary hormone excess: Genes, molecular pathways, and syndromes. Endocr. Rev. 2005, 26, 615–661. [Google Scholar] [CrossRef] [PubMed]
- Brandi, M.L.; Gagel, R.F.; Angeli, A.; Bilezikian, J.P.; Beck-Peccoz, P.; Bordi, C.; Conte-Devolx, B.; Falchetti, A.; Gheri, R.G.; Libroia, A. Consensus: Guidelines for diagnosis and therapy of MEN type 1 and type 2. J. Clin. Endocrinol. 2001, 86, 5658–5671. [Google Scholar] [CrossRef]
- Kaltsas, G.; Caplin, M.; Davies, P.; Ferone, D.; Garcia-Carbonero, R.; Grozinsky-Glasberg, S.; Horsch, D.; Tiensuu Janson, E.; Kianmanesh, R.; Kos-Kudla, B. ENETS Consensus Guidelines for the Standards of Care in Neuroendocrine Tumors: Pre- and Perioperative Therapy in Patients with Neuroendocrine Tumors. Neuroendocrinology 2017, 105, 245–254. [Google Scholar] [CrossRef]
- Baudin, E.; Caron, P.; Lombard-Bohas, C.; Tabarin, A.; Mitry, E.; Reznick, Y.; Taieb, D.; Pattou, F.; Goudet, P.; Vezzosi, D.; et al. Malignant insulinoma: Recommendations for workup and treatment. Presse Med. 2014, 43, 645–659. [Google Scholar] [CrossRef]
- Sowa-Staszczak, A.; Trofimiuk-Müldner, M.; Stefańska, A.; Tomaszuk, M.; Buziak-Bereza, M.; Gilis-Januszewska, A.; Jabrocka-Hybel, A.; Głowa, B.; Małecki, M.; Bednarczuk, T. 99mTc labeled glucagon-like peptide-1-analogue (99mTc-GLP1) scintigraphy in the management of patients with occult insulinoma. PLoS ONE 2016, 11, 1–13. [Google Scholar] [CrossRef]
- De Laat, J.M.; Dekkers, O.M.; Pieterman, C.R.C.; Kluijfhout, W.P.; Hermus, A.R.; Pereira, A.M.; van der Horst-Schrivers, A.N.; Drent, M.L.; Bisschop, P.H.; Havekes, B. Long-term natural course of pituitary tumors in patients with MEN1: Results from the Dutch MEN1 study group (DMSG). J. Clin. Endocrinol. Metab. 2015, 100, 3288–3296. [Google Scholar] [CrossRef] [Green Version]
- Lodewijk, L.; Bongers, P.J.; Kist, J.W.; Conemans, E.B.; de Laat, J.M.; Pieterman, C.R.C.; van der Horst-Schrivers, A.N.A.; Jorna, C.; Hermus, A.R.; Dekkers, O.M.; et al. Thyroid incidentalomas in patients with multiple endocrine neoplasia type 1. Eur. J. Endocrinol. 2015, 172, 337–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bademci, G.; Cengiz, F.B.; Foster, J.; Duman, D.; Sennaroglu, L.; Diaz-Horta, O.; Atik, T.; Kirazli, T.; Olgun, L.; Alper, I.; et al. Variations in Multiple Syndromic Deafness Genes Mimic Non-syndromic Hearing Loss. Sci Rep. 2016, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Feletti, A.; Anglani, M.; Scarpa, B.; Schiavi, F.; Boaretto, F.; Zovato, S.; Taschin, E.; Gardi, M.; Zanoletti, E.; Piermarocchi, S.; et al. Von Hippel-Lindau disease: An evaluation of natural history and functional disability. Neuro Oncol. 2016, 18, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerman, C.; Croyle, R.T.; Tercyak, K.P.; Hamann, H. Genetic testing: Psychological aspects and implications. J. Consult Clin. Psychol. 2002, 70, 784–797. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Sex | Age at Diagnosis | Primary Hyperparathyroidism | Kidney Stones | Pituitary Adenoma | Pancreatic Tumor | Adrenal Tumor | Other Manifestations |
|---|---|---|---|---|---|---|---|---|
| II.3 | F | 54 | Yes | Yes | No | Yes | Yes | Cutaneous collagenomas, myoma of the uterus |
| II.4 | M | 60 | Yes | Yes | No | Yes | Yes | Nodular goiter |
| III.3 | F | 35 | Yes | Yes | No | Yes | No | Cutaneous angiofibromas, nodular goiter, hypoacusia |
| III.5 | M | 34 | Yes | Yes | Yes | Yes | Yes | Cutaneous collagenomas, nodular goiter |
| III.9 | F | 23 | Yes | No | No | Yes | No | Cutaneous collagenomas |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gilis-Januszewska, A.; Bogusławska, A.; Hasse-Lazar, K.; Jurecka-Lubieniecka, B.; Jarząb, B.; Sowa-Staszczak, A.; Opalińska, M.; Godlewska, M.; Grochowska, A.; Skalniak, A.; et al. Heterogeneity of the Clinical Presentation of the MEN1 LRG_509 c.781C>T (p.Leu261Phe) Variant Within a Three-Generation Family. Genes 2021, 12, 512. https://doi.org/10.3390/genes12040512
Gilis-Januszewska A, Bogusławska A, Hasse-Lazar K, Jurecka-Lubieniecka B, Jarząb B, Sowa-Staszczak A, Opalińska M, Godlewska M, Grochowska A, Skalniak A, et al. Heterogeneity of the Clinical Presentation of the MEN1 LRG_509 c.781C>T (p.Leu261Phe) Variant Within a Three-Generation Family. Genes. 2021; 12(4):512. https://doi.org/10.3390/genes12040512
Chicago/Turabian StyleGilis-Januszewska, Aleksandra, Anna Bogusławska, Kornelia Hasse-Lazar, Beata Jurecka-Lubieniecka, Barbara Jarząb, Anna Sowa-Staszczak, Marta Opalińska, Magdalena Godlewska, Anna Grochowska, Anna Skalniak, and et al. 2021. "Heterogeneity of the Clinical Presentation of the MEN1 LRG_509 c.781C>T (p.Leu261Phe) Variant Within a Three-Generation Family" Genes 12, no. 4: 512. https://doi.org/10.3390/genes12040512
APA StyleGilis-Januszewska, A., Bogusławska, A., Hasse-Lazar, K., Jurecka-Lubieniecka, B., Jarząb, B., Sowa-Staszczak, A., Opalińska, M., Godlewska, M., Grochowska, A., Skalniak, A., & Hubalewska-Dydejczyk, A. (2021). Heterogeneity of the Clinical Presentation of the MEN1 LRG_509 c.781C>T (p.Leu261Phe) Variant Within a Three-Generation Family. Genes, 12(4), 512. https://doi.org/10.3390/genes12040512