
Krüppel-Like Factor 4 and Its Activator APTO-253 Induce NOXA-Mediated, p53-Independent Apoptosis in Triple-Negative Breast Cancer Cells

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture
2.2. Antibodies and Materials
2.3. Cell Viability Assay
2.4. Immunoblotting Analyses
2.5. RNA Interference
2.6. Quantitative Real-Time PCR (qPCR)
2.7. Promoter Assay
2.8. In Silico Analysis of Gene Expression and Kaplan–Meier Plots
2.9. UV-C Irradiation
2.10. Statistical Analysis
3. Results
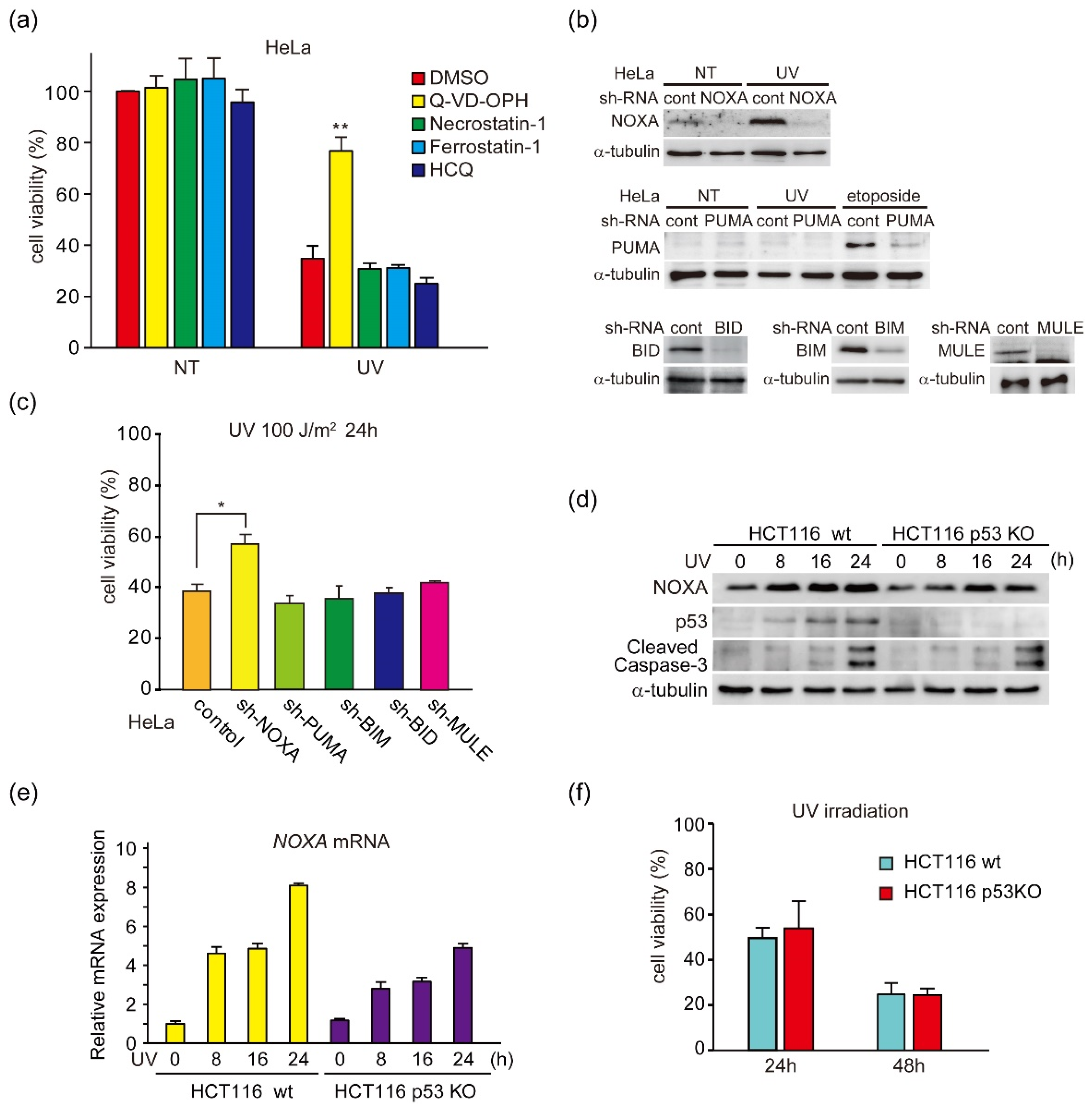
3.1. UV Light Induces p53-Independent Activation of NOXA Expression and Caspase-3
3.2. UV Light Induces NOXA Expression through the ERK Pathway
3.3. Identification of UV-Inducible Element(s) in the NOXA Gene Promoter
3.4. KLF4 Knockdown Inhibits the Induction of NOXA and Activation of Caspase-3 by UV Light
3.5. Involvement of KLF4 in the Expression of NOXA in Cancer Cells and in Survival of Breast Cancer Patients
3.6. UV-Light and Doxorubicin Induce ERK-Mediated NOXA Induction and Apoptosis in TNBC Cell Lines
3.7. KLF4 Induces Both the Expression of NOXA and Cell Death in TNBC Cell Lines
3.8. KLF4-Activating Agent APTO-253 Induces Cell Death in TNBC Cells
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbridge, A.R.; Grabow, S.; Strasser, A.; Vaux, D.L. Thirty years of BCL-2: Translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer 2016, 16, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53: 800 million years of evolution and 40 years of discovery. Nat. Rev. Cancer 2020, 20, 471–480. [Google Scholar] [CrossRef]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Hou, J.; An, Q.; Assaraf, Y.G.; Wang, X. Towards the overcoming of anticancer drug resistance mediated by p53 mutations. Drug Resist. Updates 2020, 49, 100671. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, T.; Ross, A.J.; King, A.; Zong, W.X.; Rathmell, J.C.; Shiels, H.A.; Ulrich, E.; Waymire, K.G.; Mahar, P.; Frauwirth, K.; et al. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol. Cell 2000, 6, 1389–1399. [Google Scholar] [CrossRef] [Green Version]
- Oda, E.; Ohki, R.; Murasawa, H.; Nemoto, J.; Shibue, T.; Yamashita, T.; Tokino, T.; Taniguchi, T.; Tanaka, N. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 2000, 288, 1053–1058. [Google Scholar] [CrossRef] [PubMed]
- Shibue, T.; Takeda, K.; Oda, E.; Tanaka, H.; Murasawa, H.; Takaoka, A.; Morishita, Y.; Akira, S.; Taniguchi, T.; Tanaka, N. Integral role of Noxa in p53-mediated apoptotic response. Genes Dev. 2003, 17, 2233–2238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, K.; Vousden, K.H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Yu, J.; Zhang, L.; Hwang, P.M.; Kinzler, K.W.; Vogelstein, B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol. Cell 2001, 7, 673–682. [Google Scholar] [CrossRef]
- Nakajima, W.; Tanaka, N. Synergistic induction of apoptosis by p53-inducible Bcl-2 family proteins Noxa and Puma. J. Nippon. Med. Sch. 2007, 74, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, W.; Tanaka, N. Noxa induces apoptosis in oncogene-expressing cells through catch-and-release mechanism operating between Puma and Mcl-1. Biochem. Biophys. Res. Commun. 2011, 413, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Albert, M.C.; Brinkmann, K.; Kashkar, H. Noxa and cancer therapy: Tuning up the mitochondrial death machinery in response to chemotherapy. Mol. Cell Oncol. 2014, 1, e29906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinkmann, K.; Zigrino, P.; Witt, A.; Schell, M.; Ackermann, L.; Broxtermann, P.; Schull, S.; Andree, M.; Coutelle, O.; Yazdanpanah, B.; et al. Ubiquitin C-terminal hydrolase-L1 potentiates cancer chemosensitivity by stabilizing NOXA. Cell Rep. 2013, 3, 881–891. [Google Scholar] [CrossRef] [Green Version]
- Fritsche, P.; Seidler, B.; Schuler, S.; Schnieke, A.; Gottlicher, M.; Schmid, R.M.; Saur, D.; Schneider, G. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut 2009, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakajima, W.; Sharma, K.; Hicks, M.A.; Le, N.; Brown, R.; Krystal, G.W.; Harada, H. Combination with vorinostat overcomes ABT-263 (navitoclax) resistance of small cell lung cancer. Cancer Biol. Ther. 2016, 17, 27–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torres-Adorno, A.M.; Lee, J.; Kogawa, T.; Ordentlich, P.; Tripathy, D.; Lim, B.; Ueno, N.T. Histone Deacetylase Inhibitor Enhances the Efficacy of MEK Inhibitor through NOXA-Mediated MCL1 Degradation in Triple-Negative and Inflammatory Breast Cancer. Clin. Cancer Res. 2017, 23, 4780–4792. [Google Scholar] [CrossRef] [Green Version]
- Foulkes, W.D.; Smith, I.E.; Reis-Filho, J.S. Triple-negative breast cancer. N. Engl. J. Med. 2010, 363, 1938–1948. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bykov, V.J.N.; Eriksson, S.E.; Bianchi, J.; Wiman, K.G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 2018, 18, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sionov, R.V.; Vlahopoulos, S.A.; Granot, Z. Regulation of Bim in Health and Disease. Oncotarget 2015, 6, 23058–23134. [Google Scholar] [CrossRef] [Green Version]
- Huesca, M.; Lock, L.S.; Khine, A.A.; Viau, S.; Peralta, R.; Cukier, I.H.; Jin, H.; Al-Qawasmeh, R.A.; Lee, Y.; Wright, J.; et al. A novel small molecule with potent anticancer activity inhibits cell growth by modulating intracellular labile zinc homeostasis. Mol. Cancer Ther. 2009, 8, 2586–2596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Local, A.; Zhang, H.; Benbatoul, K.D.; Folger, P.; Sheng, X.; Tsai, C.Y.; Howell, S.B.; Rice, W.G. APTO-253 Stabilizes G-quadruplex DNA, Inhibits MYC Expression, and Induces DNA Damage in Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1177–1186. [Google Scholar] [CrossRef] [Green Version]
- Kawauchi, K.; Araki, K.; Tobiume, K.; Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell Biol. 2008, 10, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Tanaka, N. IL-8-induced O-GlcNAc modification via GLUT3 and GFAT regulates cancer stem cell-like properties in colon and lung cancer cells. Oncogene 2019, 38, 1520–1533. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Wu, S.B.; Hong, C.H.; Yu, H.S.; Wei, Y.H. Molecular Mechanisms of UV-Induced Apoptosis and Its Effects on Skin Residential Cells: The Implication in UV-Based Phototherapy. Int. J. Mol. Sci. 2013, 14, 6414–6435. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Nemzow, L.; Chen, H.; Lubin, A.; Rong, X.; Sun, Z.; Harris, T.K.; Gong, F. The deubiquitinating enzyme USP24 is a regulator of the UV damage response. Cell Rep. 2015, 10, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Naik, E.; Michalak, E.M.; Villunger, A.; Adams, J.M.; Strasser, A. Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J. Cell Biol. 2007, 176, 415–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, J.Z.; Stennett, L.; Bacon, P.; Bodner, B.; Hendrix, M.J.; Seftor, R.E.; Seftor, E.A.; Margaryan, N.V.; Pollock, P.M.; Curtis, A.; et al. p53-independent NOXA induction overcomes apoptotic resistance of malignant melanomas. Mol. Cancer Ther. 2004, 3, 895–902. [Google Scholar] [PubMed]
- Polyak, K.; Waldman, T.; He, T.C.; Kinzler, K.W.; Vogelstein, B. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev. 1996, 10, 1945–1952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsen, M.; Ussat, S.; Jakob, M.; Scherer, G.; Lepenies, I.; Schutze, S.; Kabelitz, D.; Adam-Klages, S. Interaction with XIAP prevents full caspase-3/-7 activation in proliferating human T lymphocytes. Eur. J. Immunol. 2008, 38, 1979–1987. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.; Lopez, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tournier, C.; Hess, P.; Yang, D.D.; Xu, J.; Turner, T.K.; Nimnual, A.; Bar-Sagi, D.; Jones, S.N.; Flavell, R.A.; Davis, R.J. Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 2000, 288, 870–874. [Google Scholar] [CrossRef]
- Johnson, G.L.; Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science 2002, 298, 1911–1912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doudican, N.A.; Mazumder, A.; Kapoor, S.; Sultana, Z.; Kumar, A.; Talawdekar, A.; Basu, K.; Agrawal, A.; Aggarwal, A.; Shetty, K.; et al. Predictive simulation approach for designing cancer therapeutic regimens with novel biological mechanisms. J. Cancer 2014, 5, 406–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Kim, S.Y.; Song, S.J.; Hong, H.K.; Lee, Y.; Oh, B.Y.; Lee, W.Y.; Cho, Y.B. Crosstalk between CCL7 and CCR3 promotes metastasis of colon cancer cells via ERK-JNK signaling pathways. Oncotarget 2016, 7, 36842–36853. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Arechederra, M.; Sequera, C.; Bragado, P.; Vazquez-Carballo, A.; Gutierrez-Uzquiza, A.; Martin-Granado, V.; Ventura, J.J.; Kazanietz, M.G.; Guerrero, C.; et al. C3G knock-down enhances migration and invasion by increasing Rap1-mediated p38alpha activation, while it impairs tumor growth through p38alpha-independent mechanisms. Oncotarget 2016, 7, 45060–45078. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, C.; Brumatti, G.; Elgendy, M.; Brunet, M.; Martin, S.J. An ERK-dependent pathway to Noxa expression regulates apoptosis by platinum-based chemotherapeutic drugs. Oncogene 2010, 29, 6428–6441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favata, M.F.; Horiuchi, K.Y.; Manos, E.J.; Daulerio, A.J.; Stradley, D.A.; Feeser, W.S.; Van Dyk, D.E.; Pitts, W.J.; Earl, R.A.; Hobbs, F.; et al. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998, 273, 18623–18632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebolt-Leopold, J.S.; Dudley, D.T.; Herrera, R.; Van Becelaere, K.; Wiland, A.; Gowan, R.C.; Tecle, H.; Barrett, S.D.; Bridges, A.; Przybranowski, S.; et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999, 5, 810–816. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K.; Vu, T.T.; Cook, W.; Naseri, M.; Zhan, K.; Nakajima, W.; Harada, H. p53-independent Noxa induction by cisplatin is regulated by ATF3/ATF4 in head and neck squamous cell carcinoma cells. Mol. Oncol. 2018, 12, 788–798. [Google Scholar] [CrossRef]
- Luan, B.; Yoon, Y.S.; Le Lay, J.; Kaestner, K.H.; Hedrick, S.; Montminy, M. CREB pathway links PGE2 signaling with macrophage polarization. Proc. Natl. Acad. Sci. USA 2015, 112, 15642–15647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforov, M.A.; Riblett, M.; Tang, W.H.; Gratchouck, V.; Zhuang, D.; Fernandez, Y.; Verhaegen, M.; Varambally, S.; Chinnaiyan, A.M.; Jakubowiak, A.J.; et al. Tumor cell-selective regulation of NOXA by c-MYC in response to proteasome inhibition. Proc. Natl. Acad. Sci. USA 2007, 104, 19488–19493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hershko, T.; Ginsberg, D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem. 2004, 279, 8627–8634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, F.; Jin, S.; Amundson, S.A.; Tong, T.; Fan, W.; Zhao, H.; Zhu, X.; Mazzacurati, L.; Li, X.; Petrik, K.L.; et al. ATF3 induction following DNA damage is regulated by distinct signaling pathways and over-expression of ATF3 protein suppresses cells growth. Oncogene 2002, 21, 7488–7496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iordanov, M.; Bender, K.; Ade, T.; Schmid, W.; Sachsenmaier, C.; Engel, K.; Gaestel, M.; Rahmsdorf, H.J.; Herrlich, P. CREB is activated by UVC through a p38/HOG-1-dependent protein kinase. EMBO J. 1997, 16, 1009–1022. [Google Scholar] [CrossRef] [Green Version]
- Isakoff, S.J. Triple-negative breast cancer: Role of specific chemotherapy agents. Cancer J. 2010, 16, 53–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baou, M.; Kohlhaas, S.L.; Butterworth, M.; Vogler, M.; Dinsdale, D.; Walewska, R.; Majid, A.; Eldering, E.; Dyer, M.J.; Cohen, G.M. Role of NOXA and its ubiquitination in proteasome inhibitor-induced apoptosis in chronic lymphocytic leukemia cells. Haematologica 2010, 95, 1510–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghaleb, A.M.; Yang, V.W. Kruppel-like factor 4 (KLF4): What we currently know. Gene 2017, 611, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Lowman, X.H.; McDonnell, M.A.; Kosloske, A.; Odumade, O.A.; Jenness, C.; Karim, C.B.; Jemmerson, R.; Kelekar, A. The proapoptotic function of Noxa in human leukemia cells is regulated by the kinase Cdk5 and by glucose. Mol. Cell 2010, 40, 823–833. [Google Scholar] [CrossRef] [PubMed]
- Sudharsan, R.; Azuma, Y. The SUMO ligase PIAS1 regulates UV-induced apoptosis by recruiting Daxx to SUMOylated foci. J. Cell Sci. 2012, 125, 5819–5829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Regan, K.M.; Lou, Z.; Chen, J.; Tindall, D.J. CDK2-dependent phosphorylation of FOXO1 as an apoptotic response to DNA damage. Science 2006, 314, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhang, L. PUMA, a potent killer with or without p53. Oncogene 2008, 27, S71–S83. [Google Scholar] [CrossRef] [Green Version]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Villunger, A.; Michalak, E.M.; Coultas, L.; Mullauer, F.; Bock, G.; Ausserlechner, M.J.; Adams, J.M.; Strasser, A. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science 2003, 302, 1036–1038. [Google Scholar] [CrossRef] [Green Version]
- Mohan, N.; Ai, W.; Chakrabarti, M.; Banik, N.L.; Ray, S.K. KLF4 overexpression and apigenin treatment down regulated anti-apoptotic Bcl-2 proteins and matrix metalloproteinases to control growth of human malignant neuroblastoma SK-N-DZ and IMR-32 cells. Mol. Oncol. 2013, 7, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Riverso, M.; Montagnani, V.; Stecca, B. KLF4 is regulated by RAS/RAF/MEK/ERK signaling through E2F1 and promotes melanoma cell growth. Oncogene 2017, 36, 3322–3333. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Camarillo, C.; Ocampo, E.A.; Casamichana, M.L.; Perez-Plasencia, C.; Alvarez-Sanchez, E.; Marchat, L.A. Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis. Int. J. Mol. Sci 2012, 13, 142–172. [Google Scholar] [CrossRef]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.F.; Nebreda, A.R. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 2009, 9, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Cagnol, S.; Chambard, J.C. ERK and cell death: Mechanisms of ERK-induced cell death--apoptosis, autophagy and senescence. FEBS J. 2010, 277, 2–21. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Dai, W.; Lu, L. Ultraviolet-induced junD activation and apoptosis in myeloblastic leukemia ML-1 cells. J. Biol. Chem. 2002, 277, 32668–32676. [Google Scholar] [CrossRef] [Green Version]
- Dhaliwal, N.K.; Miri, K.; Davidson, S.; Tamim El Jarkass, H.; Mitchell, J.A. KLF4 Nuclear Export Requires ERK Activation and Initiates Exit from Naive Pluripotency. Stem Cell Rep. 2018, 10, 1308–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 2016, 17, 183–193. [Google Scholar] [CrossRef]
- McConnell, B.B.; Yang, V.W. Mammalian Kruppel-like factors in health and diseases. Physiol. Rev. 2010, 90, 1337–1381. [Google Scholar] [CrossRef]
- Qi, X.T.; Li, Y.L.; Zhang, Y.Q.; Xu, T.; Lu, B.; Fang, L.; Gao, J.Q.; Yu, L.S.; Zhu, D.F.; Yang, B.; et al. KLF4 functions as an oncogene in promoting cancer stem cell-like characteristics in osteosarcoma cells. Acta Pharmacol. Sin. 2019, 40, 546–555. [Google Scholar] [CrossRef]
- Rowland, B.D.; Bernards, R.; Peeper, D.S. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat. Cell Biol. 2005, 7, 1074–1082. [Google Scholar] [CrossRef]
- Wang, B.; Zhao, M.Z.; Cui, N.P.; Lin, D.D.; Zhang, A.Y.; Qin, Y.; Liu, C.Y.; Yan, W.T.; Shi, J.H.; Chen, B.P. Kruppel-like factor 4 induces apoptosis and inhibits tumorigenic progression in SK-BR-3 breast cancer cells. FEBS Open Bio 2015, 5, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowland, B.D.; Peeper, D.S. KLF4, p21 and context-dependent opposing forces in cancer. Nat. Rev. Cancer 2006, 6, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Wheler, J.; Murray, P.E.; Zhou, S.; Saltz, L. Phase 1 study of APTO-253 HCl, an inducer of KLF4, in patients with advanced or metastatic solid tumors. Investig. New Drugs 2015, 33, 1086–1092. [Google Scholar] [CrossRef]
- Blows, F.M.; Driver, K.E.; Schmidt, M.K.; Broeks, A.; van Leeuwen, F.E.; Wesseling, J.; Cheang, M.C.; Gelmon, K.; Nielsen, T.O.; Blomqvist, C.; et al. Subtyping of breast cancer by immunohistochemistry to investigate a relationship between subtype and short and long term survival: A collaborative analysis of data for 10,159 cases from 12 studies. PLoS Med. 2010, 7, e1000279. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.; Winer, E.; Viale, G.; Cameron, D.; Gianni, L. Triple-negative breast cancer: Disease entity or title of convenience? Nat. Rev. Clin. Oncol. 2010, 7, 683–692. [Google Scholar] [CrossRef]
- Gupta, G.K.; Collier, A.L.; Lee, D.; Hoefer, R.A.; Zheleva, V.; Siewertsz van Reesema, L.L.; Tang-Tan, A.M.; Guye, M.L.; Chang, D.Z.; Winston, J.S.; et al. Perspectives on Triple-Negative Breast Cancer: Current Treatment Strategies, Unmet Needs, and Potential Targets for Future Therapies. Cancers 2020, 12, 2392. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakajima, W.; Miyazaki, K.; Asano, Y.; Kubota, S.; Tanaka, N. Krüppel-Like Factor 4 and Its Activator APTO-253 Induce NOXA-Mediated, p53-Independent Apoptosis in Triple-Negative Breast Cancer Cells. Genes 2021, 12, 539. https://doi.org/10.3390/genes12040539
Nakajima W, Miyazaki K, Asano Y, Kubota S, Tanaka N. Krüppel-Like Factor 4 and Its Activator APTO-253 Induce NOXA-Mediated, p53-Independent Apoptosis in Triple-Negative Breast Cancer Cells. Genes. 2021; 12(4):539. https://doi.org/10.3390/genes12040539
Chicago/Turabian StyleNakajima, Wataru, Kai Miyazaki, Yumi Asano, Satoshi Kubota, and Nobuyuki Tanaka. 2021. "Krüppel-Like Factor 4 and Its Activator APTO-253 Induce NOXA-Mediated, p53-Independent Apoptosis in Triple-Negative Breast Cancer Cells" Genes 12, no. 4: 539. https://doi.org/10.3390/genes12040539