
Generating New FANCA-Deficient HNSCC Cell Lines by Genomic Editing Recapitulates the Cellular Phenotypes of Fanconi Anemia

 , , ,
, , ,  ,
,
Abstract
:1. Introduction
2. Results
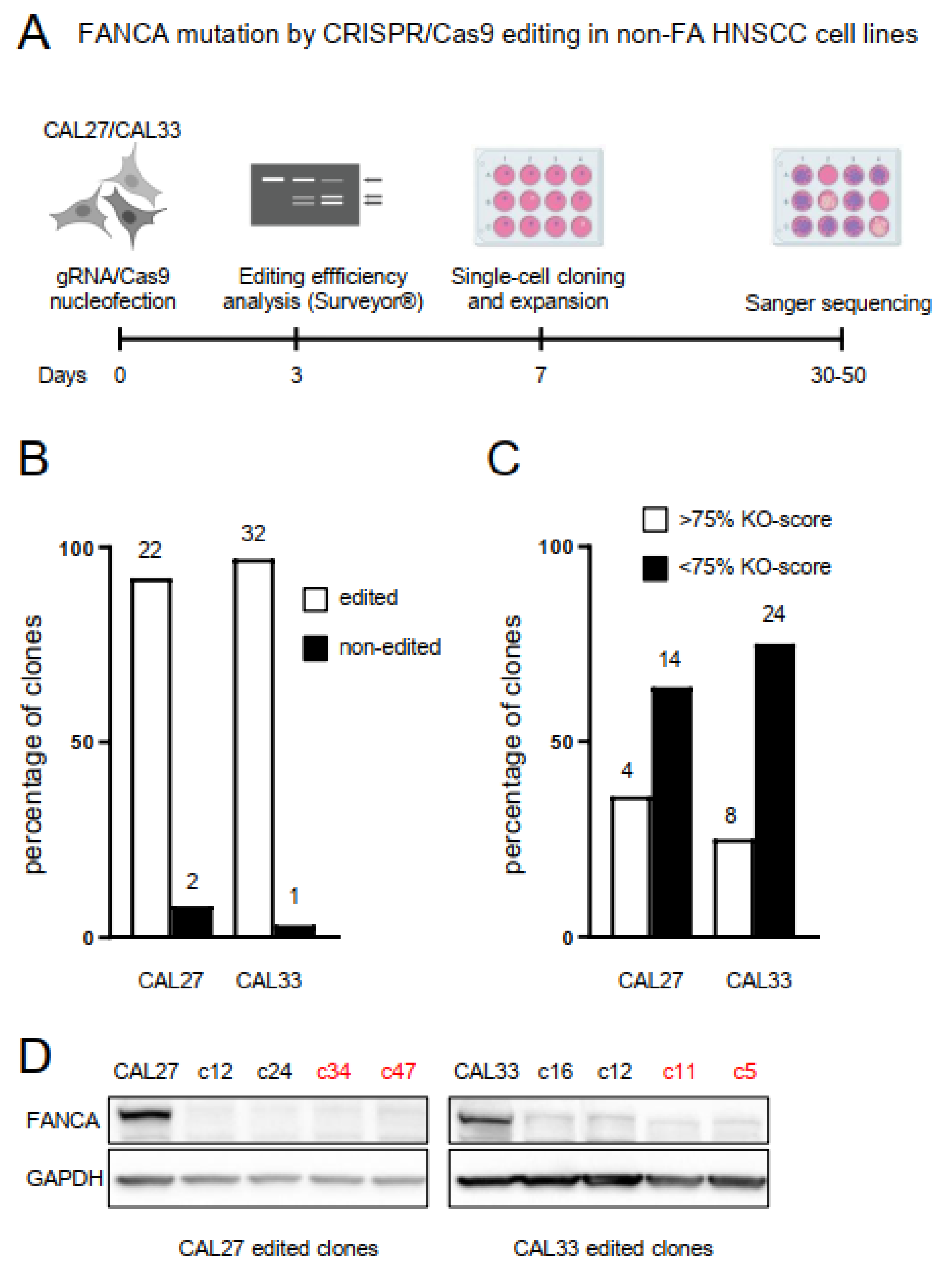
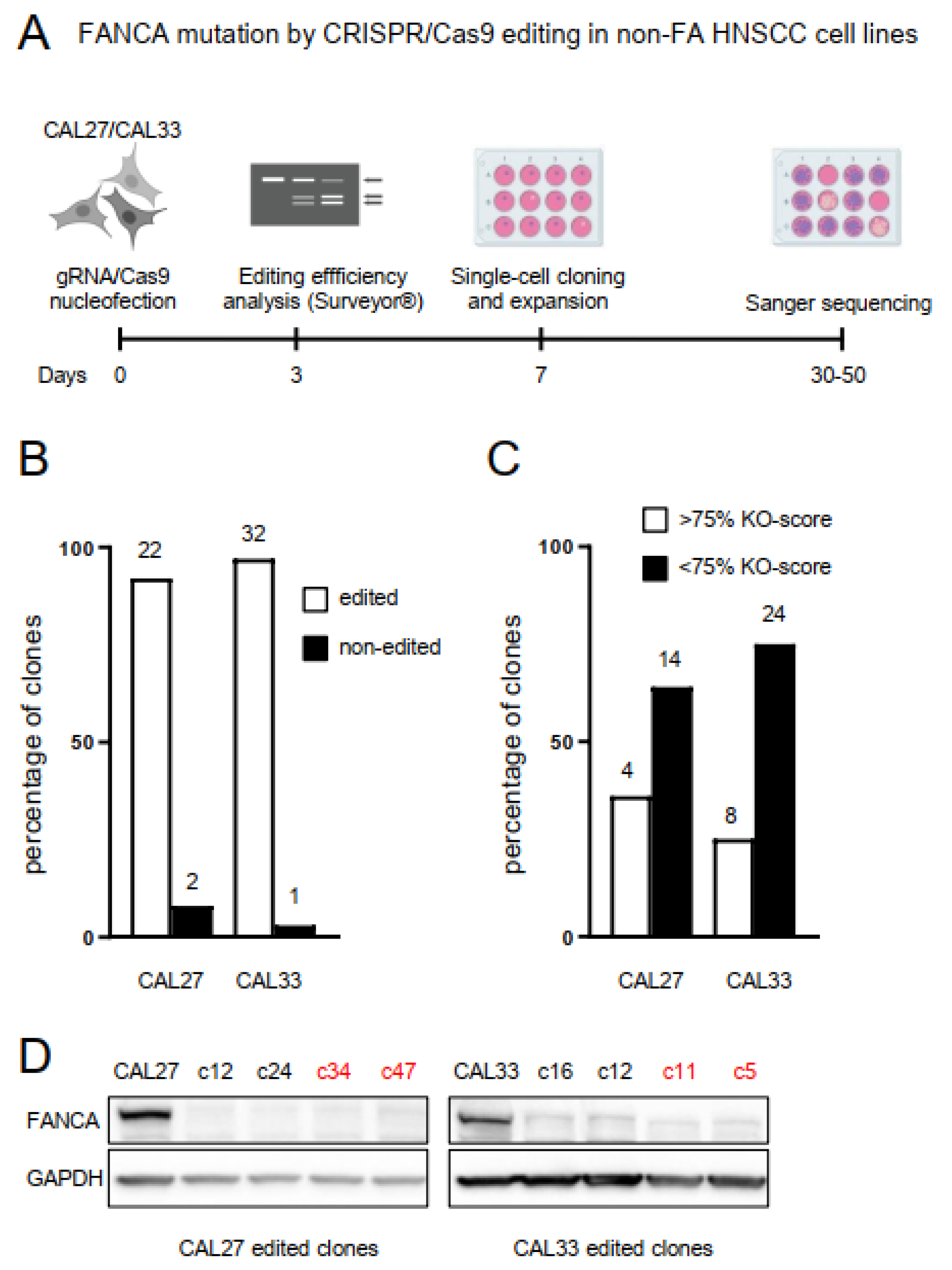
2.1. Biallelic Mutation in the FANCA Gene by CRISPR/Cas9 Editing in Non-FA HNSCC Cell Lines
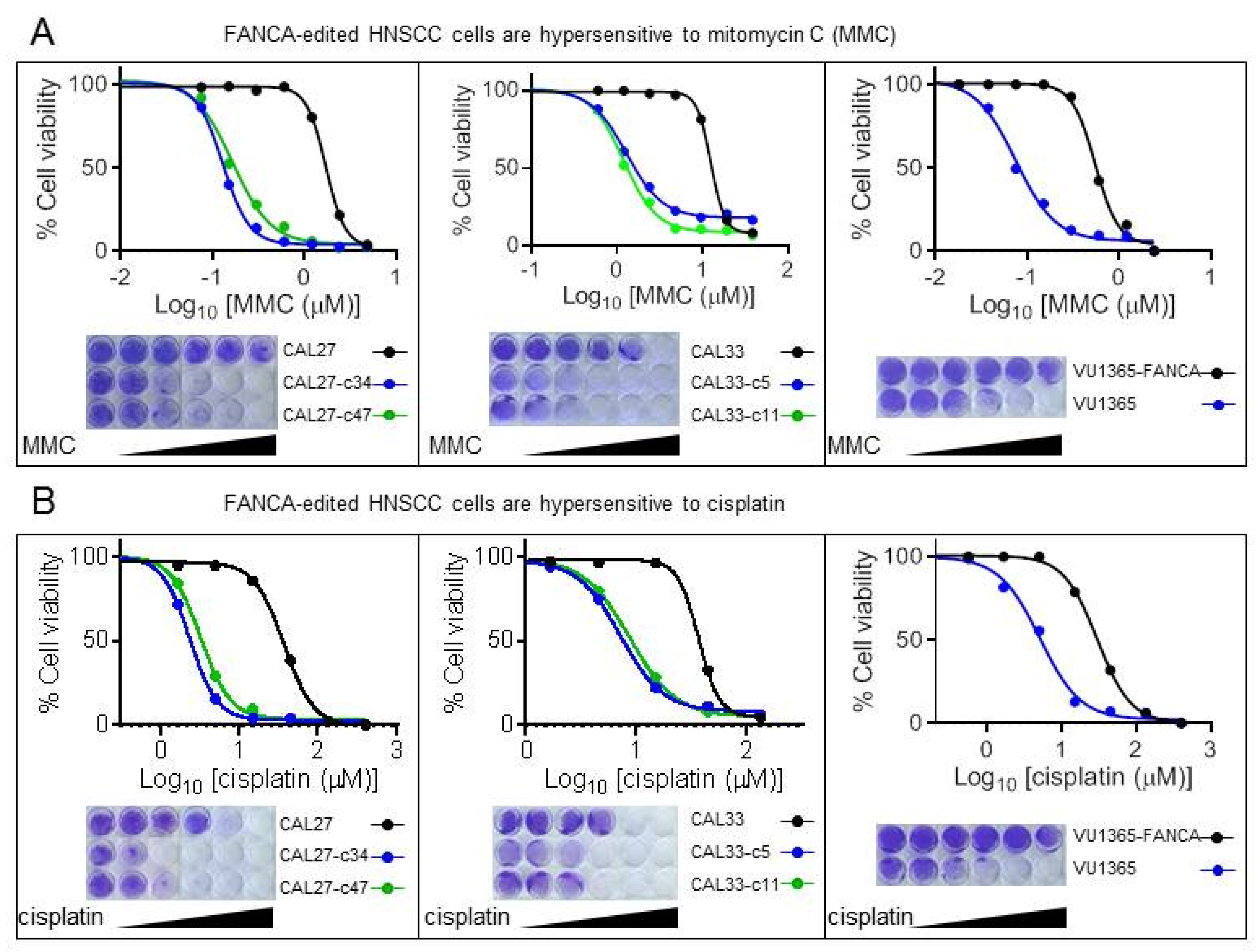
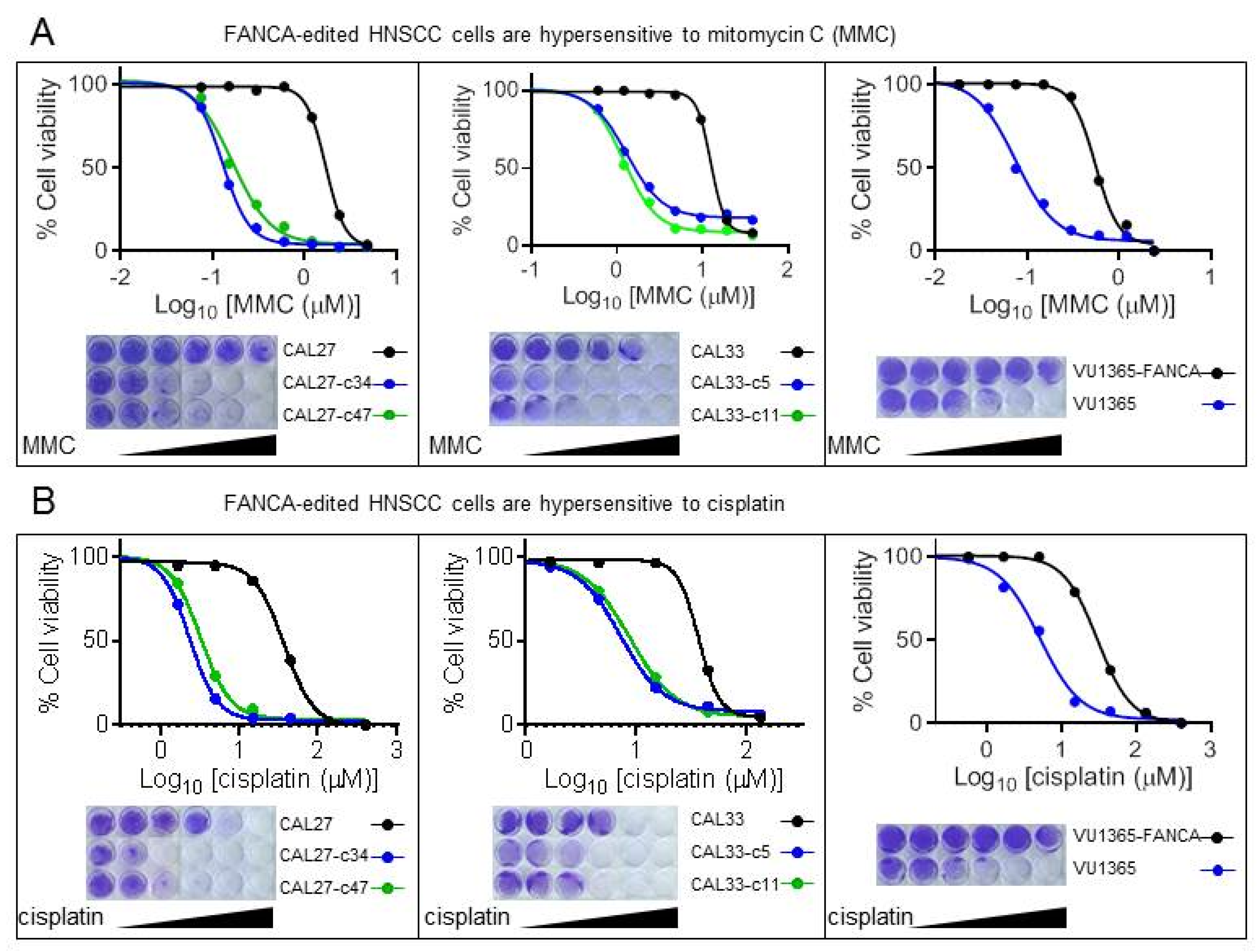
2.2. FANCA-Mutant Clones Are Hypersensitive to ICL-Agents
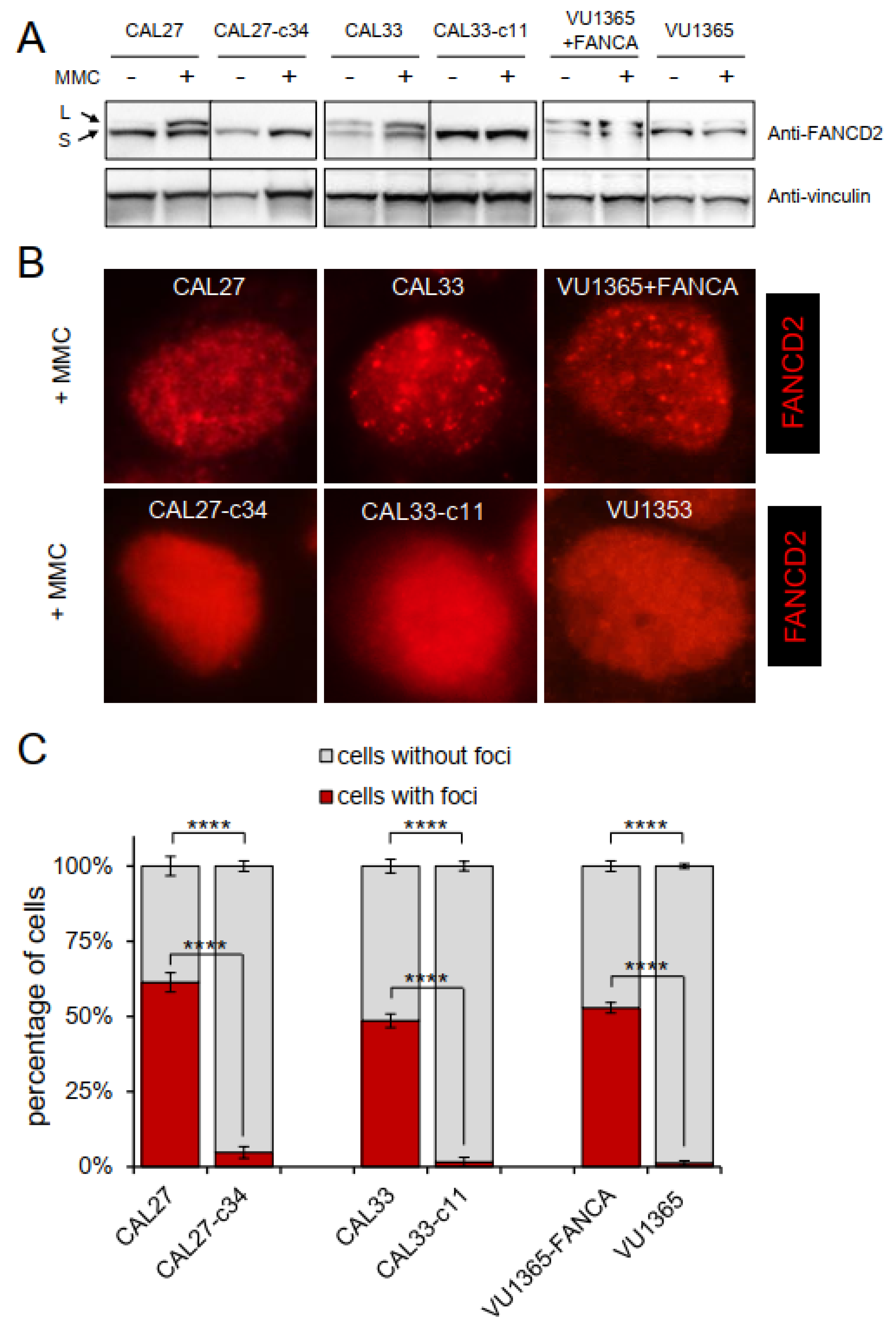
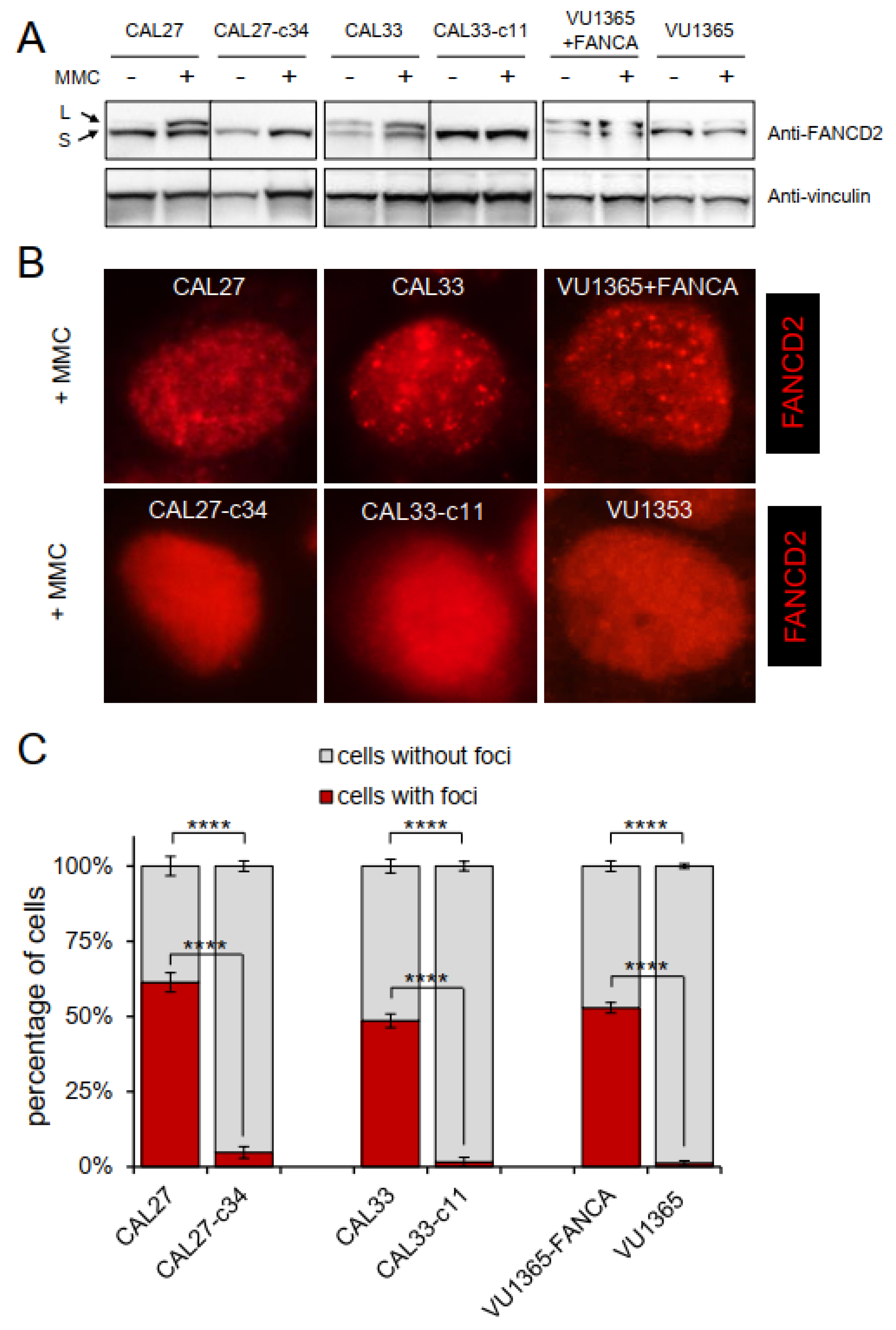
2.3. FANCA-Mutant Clones Are Defective in FANCD2 Monoubiquitination and Nuclear Foci Formation
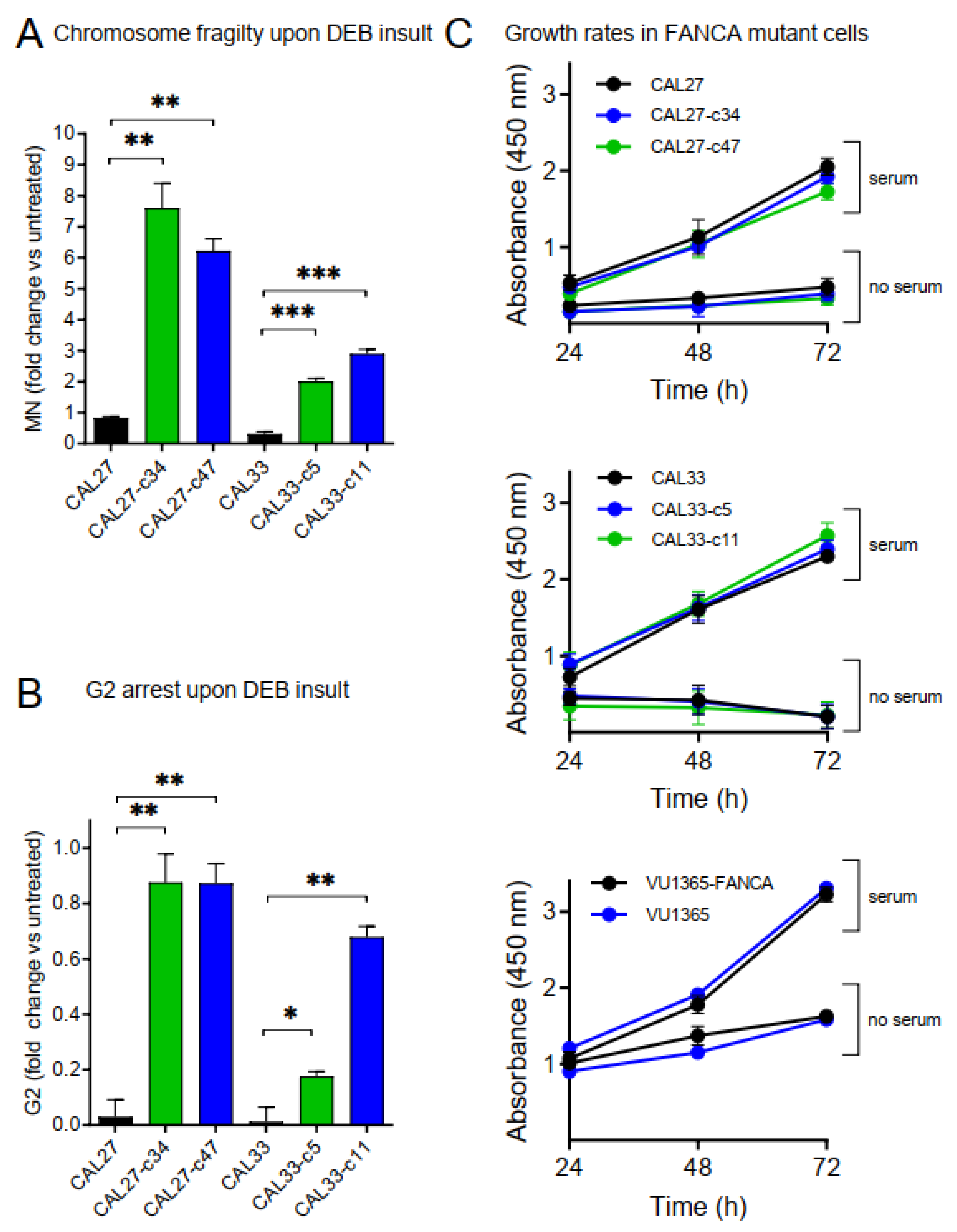
2.4. Increased Chromosome Fragility and G2 Arrest in FANCA-Mutant Clones
2.5. FANCA-Mutation Does Not Affect Growth Rates in HNSCC Cells
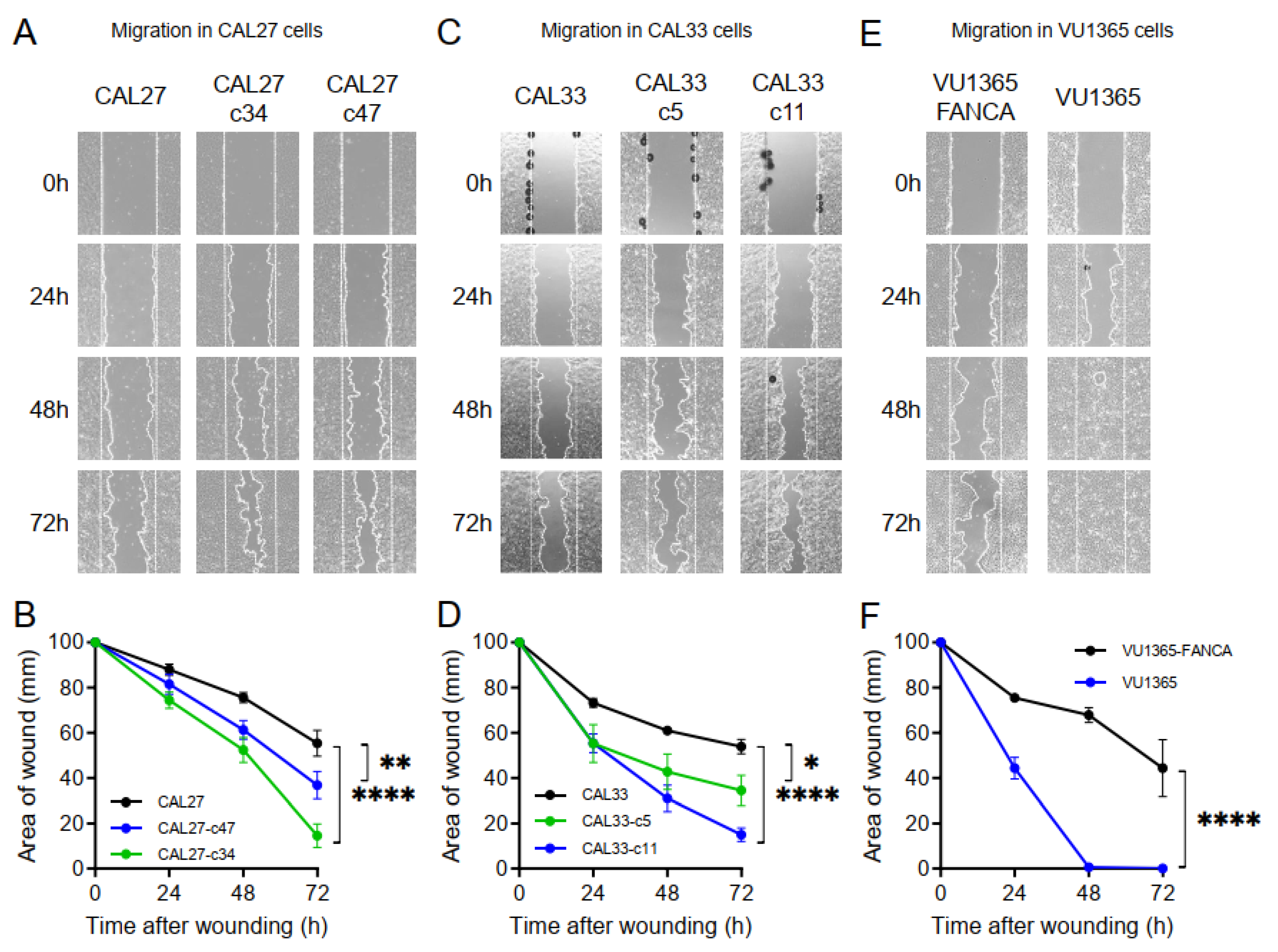
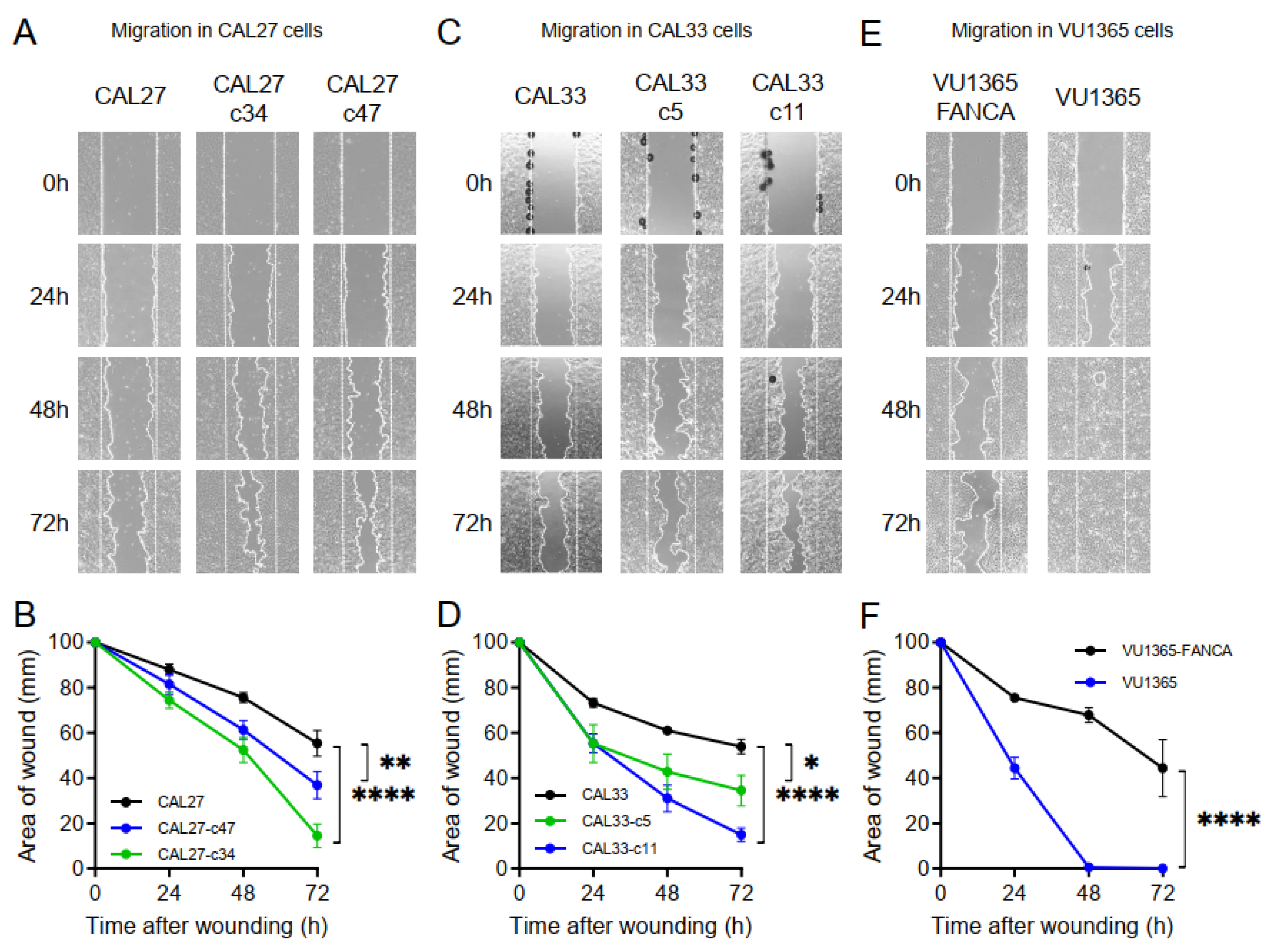
2.6. FANCA-Mutation Augments Cell Migration in HNSCC Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Genotoxic Agents
4.2. Cell Line Nucleofection and CRISPR/Cas9 Editing within Exon 4 of FANCA Gene
4.3. Protein Extraction and Western Blotting
4.4. Mitomycin C (MMC) and Cisplatin Sensitivity Assays
4.5. FANCD2 Immunofluorescence
4.6. Chromosome Fragility and G2 Arrest by the Flow Cytometry Micronucleus (MN) Test
4.7. Cell Growth Assays
4.8. Wound Healing Assay for Cell Migration Analysis
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bogliolo, M.; Surralles, J. Fanconi anemia: A model disease for studies on human genetics and advanced therapeutics. Curr. Opin. Genet. Dev. 2015, 33, 32–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niraj, J.; Farkkila, A.; D’andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Rio, P.; Navarro, S.; Wang, W.; Sanchez-Dominguez, R.; Pujol, R.M.; Segovia, J.C.; Bogliolo, M.; Merino, E.; Wu, N.; Salgado, R.; et al. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019, 25, 1396–1401. [Google Scholar] [CrossRef]
- Risitano, A.M.; Marotta, S.; Calzone, R.; Grimaldi, F.; Zatterale, A.; Contributors, R. Twenty years of the Italian Fanconi Anemia Registry: Where we stand and what remains to be learned. Haematologica 2016, 101, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alter, B.P.; Giri, N.; Savage, S.A.; Rosenberg, P.S. Cancer in the National Cancer Institute inherited bone marrow failure syndrome cohort after fifteen years of follow-up. Haematologica 2018, 103, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi anemia links reactive oxygen species to insulin resistance and obesity. Antioxid. Redox. Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef]
- Zhang, Q.S.; Tang, W.; Deater, M.; Phan, N.; Marcogliese, A.N.; Li, H.; Al-Dhalimy, M.; Major, A.; Olson, S.; Monnat, R.J., Jr.; et al. Metformin improves defective hematopoiesis and delays tumor formation in Fanconi anemia mice. Blood 2016, 128, 2774–2784. [Google Scholar] [CrossRef] [Green Version]
- Kutler, D.I.; Patel, K.R.; Auerbach, A.D.; Kennedy, J.; Lach, F.P.; Sanborn, E.; Cohen, M.A.; Kuhel, W.I.; Smogorzewska, A. Natural history and management of Fanconi anemia patients with head and neck cancer: A 10-year follow-up. Laryngoscope 2016, 126, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Montanuy, H.; Martinez-Barriocanal, A.; Casado, J.A.; Rovirosa, L.; Ramirez, M.J.; Nieto, R.; Carrascoso-Rubio, C.; Riera, P.; Gonzalez, A.; Lerma, E.; et al. Gefitinib and afatinib show potential efficacy for Fanconi anemia-related head and neck cancer. Clin. Cancer Res. 2020, 26, 3044–3057. [Google Scholar] [CrossRef] [Green Version]
- Van Harten, A.M.; Poell, J.B.; Buijze, M.; Brink, A.; Wells, S.I.; Rene Leemans, C.; Wolthuis, R.M.F.; Brakenhoff, R.H. Characterization of a head and neck cancer-derived cell line panel confirms the distinct TP53-proficient copy number-silent subclass. Oral Oncol. 2019, 98, 53–61. [Google Scholar] [CrossRef]
- Van Zeeburg, H.J.; Snijders, P.J.; Pals, G.; Hermsen, M.A.; Rooimans, M.A.; Bagby, G.; Soulier, J.; Gluckman, E.; Wennerberg, J.; Leemans, C.R.; et al. Generation and molecular characterization of head and neck squamous cell lines of fanconi anemia patients. Cancer Res. 2005, 65, 1271–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; Mcdonald, E.R.; 3rd Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Abba, M.C.; Molinolo, A.A.; Vitale-Cross, L.; Wang, Z.; Zaida, M.; Delic, N.C.; Samuels, Y.; Lyons, J.G.; Gutkind, J.S. The head and neck cancer cell oncogenome: A platform for the development of precision molecular therapies. Oncotarget 2014, 5, 8906–8923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Palacios-Garcia, J.; Sanz-Flores, M.; Asensio, A.; Alvarado, R.; Rojo-Berciano, S.; Stamatakis, K.; Paramio, J.M.; Cano, A.; Nieto, M.A.; Garcia-Escudero, R.; et al. G-protein-coupled receptor kinase 2 safeguards epithelial phenotype in head and neck squamous cell carcinomas. Int. J. Cancer 2019, 147, 218–229. [Google Scholar] [CrossRef]
- Hsiau, T.; Conant, D.; Rossi, N.; Maures, T.; Waite, K.; Yang, J.; Joshi, S.; Kelso, R.; Holden, K.; Enzmann, B.L.; et al. Inference of CRISPR Edits from Sanger Trace Data. bioRxiv 2019. [Google Scholar] [CrossRef]
- Giampietro, P.F.; Adler-Brecher, B.; Verlander, P.C.; Pavlakis, S.G.; Davis, J.G.; Auerbach, A.D. The need for more accurate and timely diagnosis in Fanconi anemia: A report from the International Fanconi Anemia Registry. Pediatrics 1993, 91, 1116–1120. [Google Scholar]
- Auerbach, A.D. Fanconi anemia diagnosis and the diepoxybutane (DEB) test. Exp. Hematol. 1993, 21, 731–733. [Google Scholar]
- Garcia-Higuera, I.; Taniguchi, T.; Ganesan, S.; Meyn, M.S.; Timmers, C.; Hejna, J.; Grompe, M.; D’andrea, A.D. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell. 2001, 7, 249–262. [Google Scholar] [CrossRef]
- Lombardi, A.J.; Hoskins, E.E.; Foglesong, G.D.; Wikenheiser-Brokamp, K.A.; Wiesmuller, L.; Hanenberg, H.; Andreassen, P.R.; Jacobs, A.J.; Olson, S.B.; Keeble, W.W.; et al. Acquisition of Relative Interstrand Crosslinker Resistance and PARP Inhibitor Sensitivity in Fanconi Anemia Head and Neck Cancers. Clin. Cancer Res. 2015, 21, 1962–1972. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, M.J.; Minguillon, J.; Loveless, S.; Lake, K.; Carrasco, E.; Stjepanovic, N.; Balmana, J.; Catala, A.; Mehta, P.A.; Surralles, J. Chromosome fragility in the buccal epithelium in patients with Fanconi anemia. Cancer Lett. 2020, 472, 1–7. [Google Scholar] [CrossRef]
- Chandra, S.; Levran, O.; Jurickova, I.; Maas, C.; Kapur, R.; Schindler, D.; Henry, R.; Milton, K.; Batish, S.D.; Cancelas, J.A.; et al. A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol. Ther. 2005, 12, 976–984. [Google Scholar] [CrossRef]
- Rio, P.; Navarro, S.; Guenechea, G.; Sanchez-Dominguez, R.; Lamana, M.L.; Yanez, R.; Casado, J.A.; Mehta, P.A.; Pujol, M.R.; Surralles, J.; et al. Engraftment and in vivo proliferation advantage of gene-corrected mobilized CD34(+) cells from Fanconi anemia patients. Blood 2017, 130, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Sii-Felice, K.; Etienne, O.; Hoffschir, F.; Mathieu, C.; Riou, L.; Barroca, V.; Haton, C.; Arwert, F.; Fouchet, P.; Boussin, F.D.; et al. Fanconi DNA repair pathway is required for survival and long-term maintenance of neural progenitors. EMBO J. 2008, 27, 770–781. [Google Scholar] [CrossRef] [Green Version]
- Romick-Rosendale, L.E.; Hoskins, E.E.; Privette Vinnedge, L.M.; Foglesong, G.D.; Brusadelli, M.G.; Potter, S.S.; Komurov, K.; Brugmann, S.A.; Lambert, P.F.; Kimple, R.J.; et al. Defects in the Fanconi Anemia Pathway in Head and Neck Cancer Cells Stimulate Tumor Cell Invasion through DNA-PK and Rac1 Signaling. Clin. Cancer Res. 2016, 22, 2062–2073. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Martin, D.; Molinolo, A.A.; Patel, V.; Iglesias-Bartolome, R.; Degese, M.S.; Vitale-Cross, L.; Chen, Q.; Gutkind, J.S. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.K.; Jang, K.W.; Kim, S.; Kim, H.S.; Kim, S.M.; Kwon, H.J.; Kim, H.R.; Yun, H.J.; Ahn, M.J.; Park, K.U.; et al. Exome sequencing reveals recurrent REV3L mutations in cisplatin-resistant squamous cell carcinoma of head and neck. Sci. Rep. 2016, 6, 19552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eke, I.; Deuse, Y.; Hehlgans, S.; Gurtner, K.; Krause, M.; Baumann, M.; Shevchenko, A.; Sandfort, V.; Cordes, N. β1 Integrin/FAK/cortactin signaling is essential for human head and neck cancer resistance to radiotherapy. J. Clin. Invest. 2012, 122, 1529–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garnett, M.J.; Edelman, E.J.; Heidorn, S.J.; Greenman, C.D.; Dastur, A.; Lau, K.W.; Greninger, P.; Thompson, I.R.; Luo, X.; Soares, J.; et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 2012, 483, 570–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoskins, E.E.; Gunawardena, R.W.; Habash, K.B.; Wise-Draper, T.M.; Jansen, M.; Knudsen, E.S.; Wells, S.I. Coordinate regulation of Fanconi anemia gene expression occurs through the Rb/E2F pathway. Oncogene 2008, 27, 4798–4808. [Google Scholar] [CrossRef] [Green Version]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.K.; Taunk, N.K.; et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhide, S.A.; Thway, K.; Lee, J.; Wong, K.; Clarke, P.; Newbold, K.L.; Nutting, C.M.; Harrington, K.J. Delayed DNA double-strand break repair following platin-based chemotherapy predicts treatment response in head and neck squamous cell carcinoma. Br. J. Cancer 2016, 115, 825–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Essers, P.B.M.; Van Der Heijden, M.; Verhagen, C.V.M.; Ploeg, E.M.; De Roest, R.H.; Leemans, C.R.; Brakenhoff, R.H.; Van Den Brekel, M.W.M.; Bartelink, H.; Verheij, M.; et al. Drug Sensitivity Prediction Models Reveal a Link between DNA Repair Defects and Poor Prognosis in HNSCC. Cancer Res. 2019, 79, 5597–5611. [Google Scholar] [CrossRef] [Green Version]
- Antonio Casado, J.; Callen, E.; Jacome, A.; Rio, P.; Castella, M.; Lobitz, S.; Ferro, T.; Munoz, A.; Sevilla, J.; Cantalejo, A.; et al. A comprehensive strategy for the subtyping of patients with Fanconi anaemia: Conclusions from the Spanish Fanconi Anemia Research Network. J. Med. Genet. 2007, 44, 241–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avlasevich, S.L.; Bryce, S.M.; Cairns, S.E.; Dertinger, S.D. In vitro micronucleus scoring by flow cytometry: Differential staining of micronuclei versus apoptotic and necrotic chromatin enhances assay reliability. Environ. Mol. Mutagen. 2006, 47, 56–66. [Google Scholar] [CrossRef]
- Hernandez, G.; Ramirez, M.J.; Minguillon, J.; Quiles, P.; Ruiz De Garibay, G.; Aza-Carmona, M.; Bogliolo, M.; Pujol, R.; Prados-Carvajal, R.; Fernandez, J.; et al. Decapping protein EDC4 regulates DNA repair and phenocopies BRCA1. Nat. Commun. 2018, 9, 967. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | IC50 MMC | Ratio Mutant/Non Mutant | IC50 Cisplatin | Ratio Mutant/Non Mutant |
|---|---|---|---|---|
| CAL27 | 1.704 | 37.560 | ||
| CAL27-c34 | 0.129 | 13.220 | 2.377 | 15.801 |
| CAL27-c47 | 0.165 | 10.340 | 3.274 | 11.472 |
| CAL33 | 12.490 | 37.130 | ||
| CAL33-c5 | 1.298 | 9.622 | 7.281 | 5.100 |
| CAL33-c11 | 1.198 | 10.426 | 8.331 | 4.457 |
| VU1365-FANCA | 0.548 | 29.680 | ||
| VU1365 | 0.074 | 7.363 | 5.017 | 5.916 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Errazquin, R.; Sieiro, E.; Moreno, P.; Ramirez, M.J.; Lorz, C.; Peral, J.; Ortiz, J.; Casado, J.A.; Roman-Rodriguez, F.J.; Hanenberg, H.; et al. Generating New FANCA-Deficient HNSCC Cell Lines by Genomic Editing Recapitulates the Cellular Phenotypes of Fanconi Anemia. Genes 2021, 12, 548. https://doi.org/10.3390/genes12040548
Errazquin R, Sieiro E, Moreno P, Ramirez MJ, Lorz C, Peral J, Ortiz J, Casado JA, Roman-Rodriguez FJ, Hanenberg H, et al. Generating New FANCA-Deficient HNSCC Cell Lines by Genomic Editing Recapitulates the Cellular Phenotypes of Fanconi Anemia. Genes. 2021; 12(4):548. https://doi.org/10.3390/genes12040548
Chicago/Turabian StyleErrazquin, Ricardo, Esther Sieiro, Pilar Moreno, María José Ramirez, Corina Lorz, Jorge Peral, Jessica Ortiz, José Antonio Casado, Francisco J. Roman-Rodriguez, Helmut Hanenberg, and et al. 2021. "Generating New FANCA-Deficient HNSCC Cell Lines by Genomic Editing Recapitulates the Cellular Phenotypes of Fanconi Anemia" Genes 12, no. 4: 548. https://doi.org/10.3390/genes12040548
APA StyleErrazquin, R., Sieiro, E., Moreno, P., Ramirez, M. J., Lorz, C., Peral, J., Ortiz, J., Casado, J. A., Roman-Rodriguez, F. J., Hanenberg, H., Río, P., Surralles, J., Segrelles, C., & Garcia-Escudero, R. (2021). Generating New FANCA-Deficient HNSCC Cell Lines by Genomic Editing Recapitulates the Cellular Phenotypes of Fanconi Anemia. Genes, 12(4), 548. https://doi.org/10.3390/genes12040548