
Correlation Networks Provide New Insights into the Architecture of Testicular Steroid Pathways in Pigs

 ,
,  , and
, and 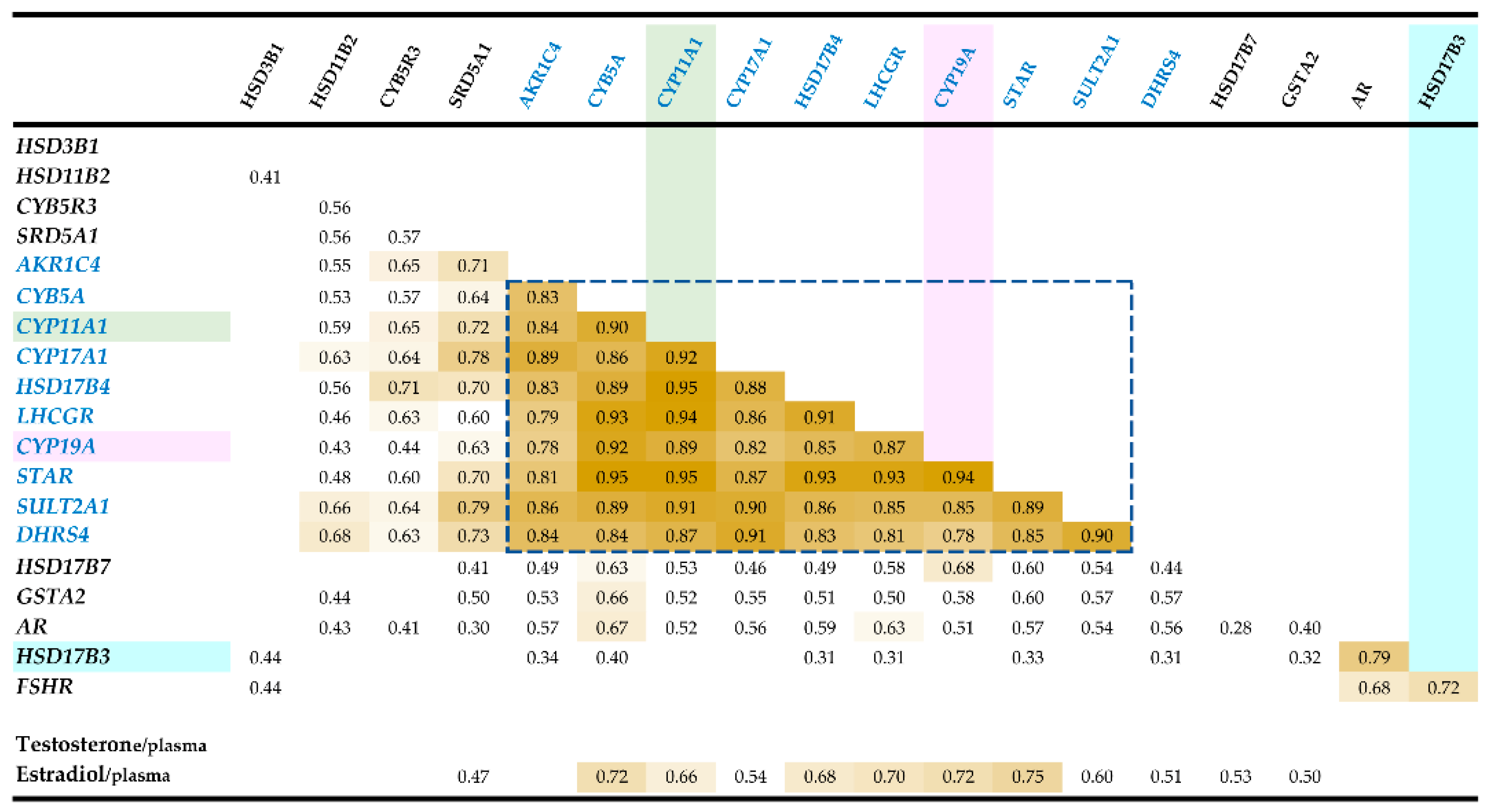{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals, Sample Collection
2.2. Gene Expression Analysis by RNA-Seq
2.3. Gene Expression Analysis by RTqPCR
2.4. Expression Quantification of Genes Localized in Genomic Clusters (CYP19A, AKR1C and SULT1C)
2.5. Statistical Analyses
2.6. Functional Enrichment Analyses
3. Results
3.1. RT-qPCR-Correlation Analyses
3.2. Detection of Correlation Groups in mRNAseq
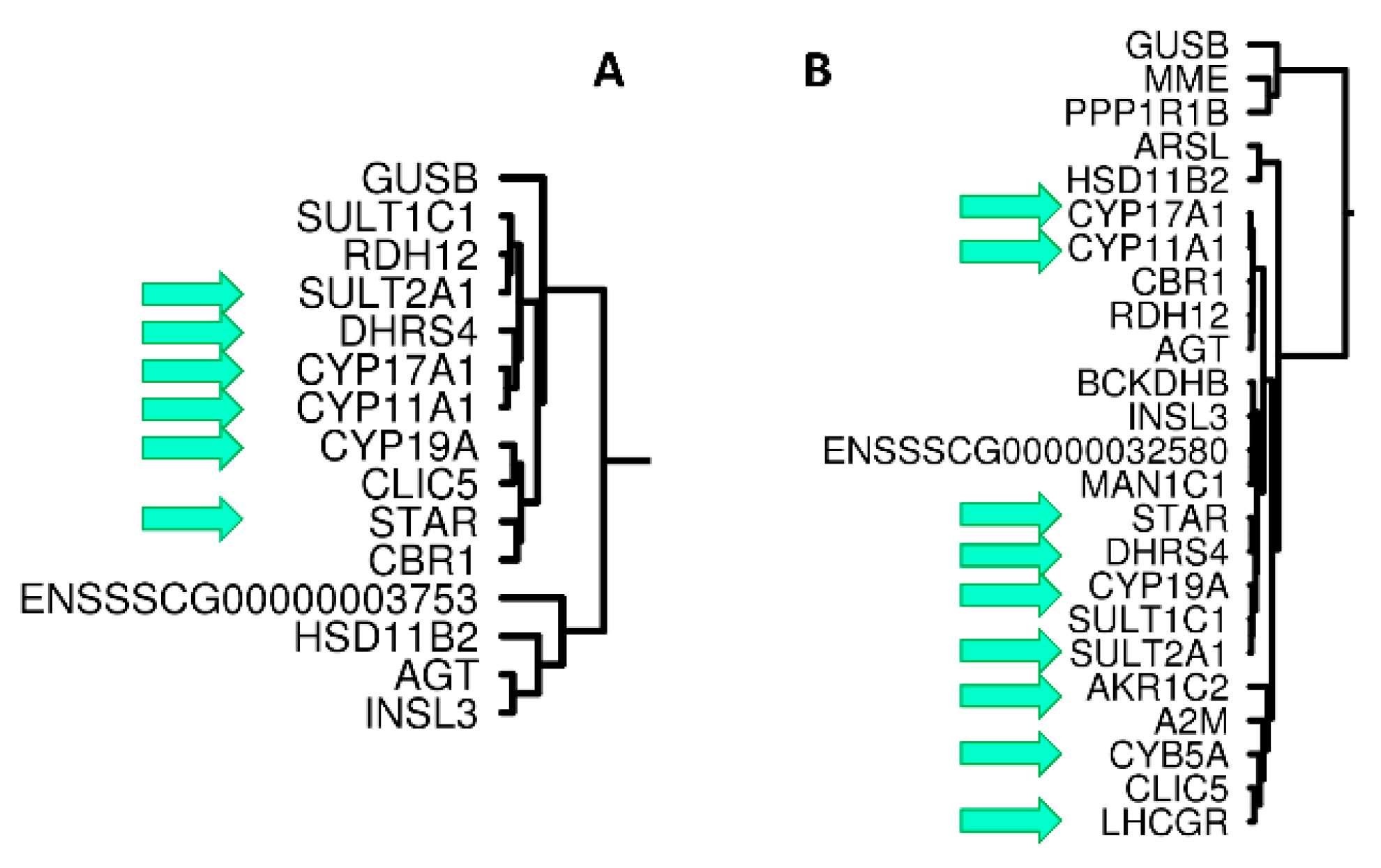
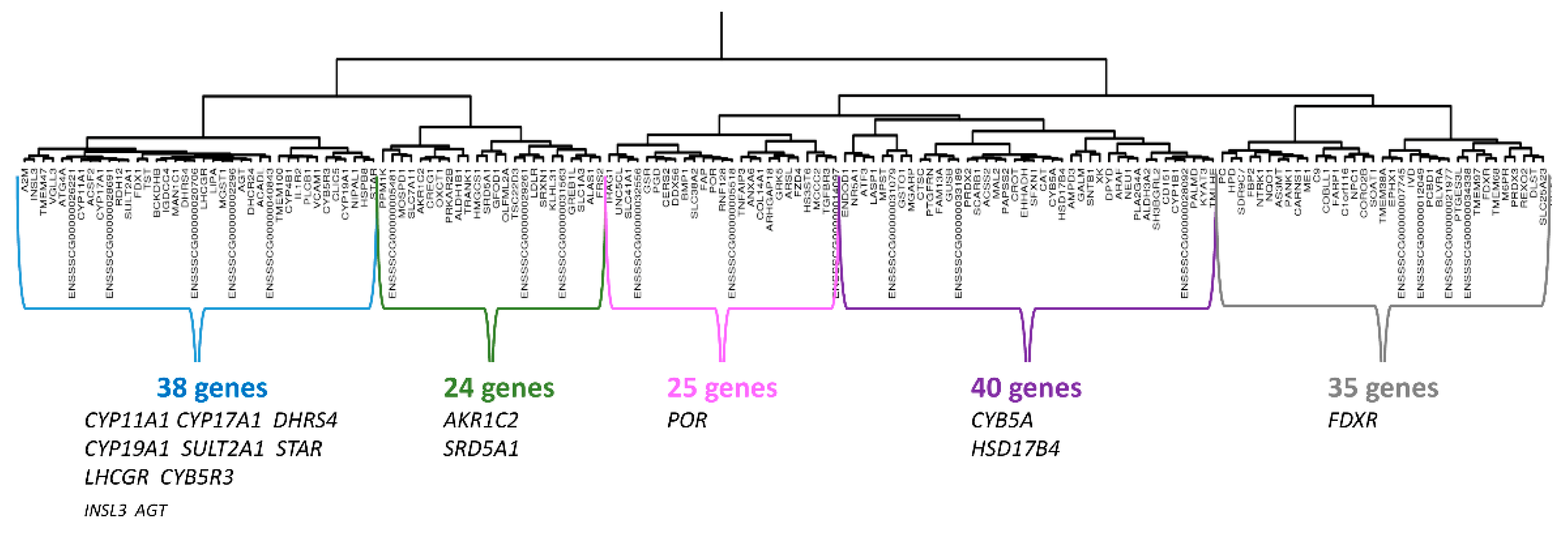
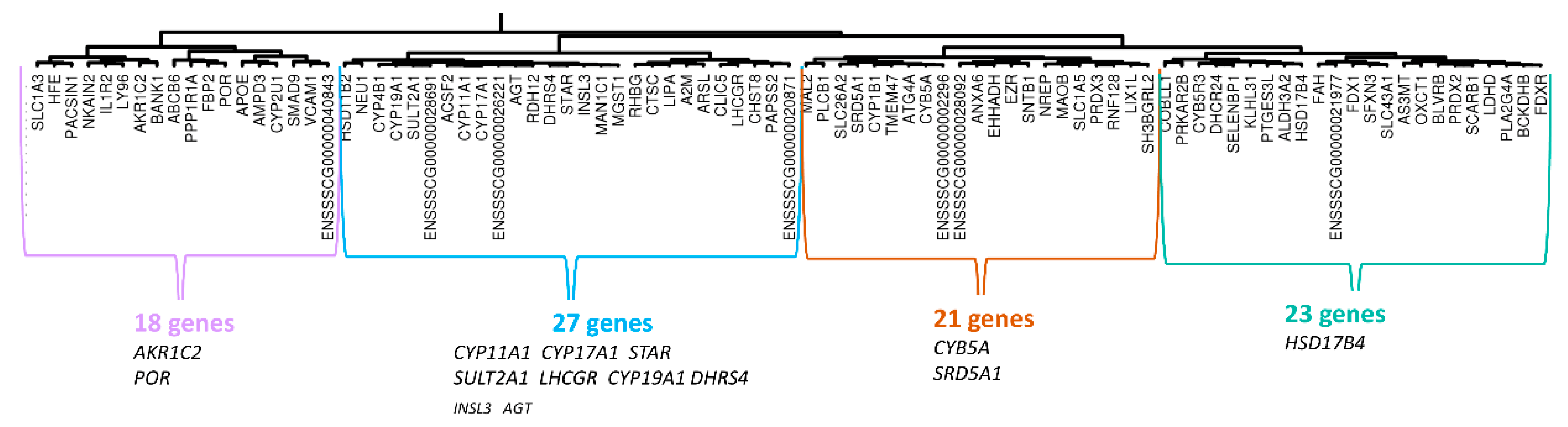
3.2.1. Hierarchical Clustering mRNAseq
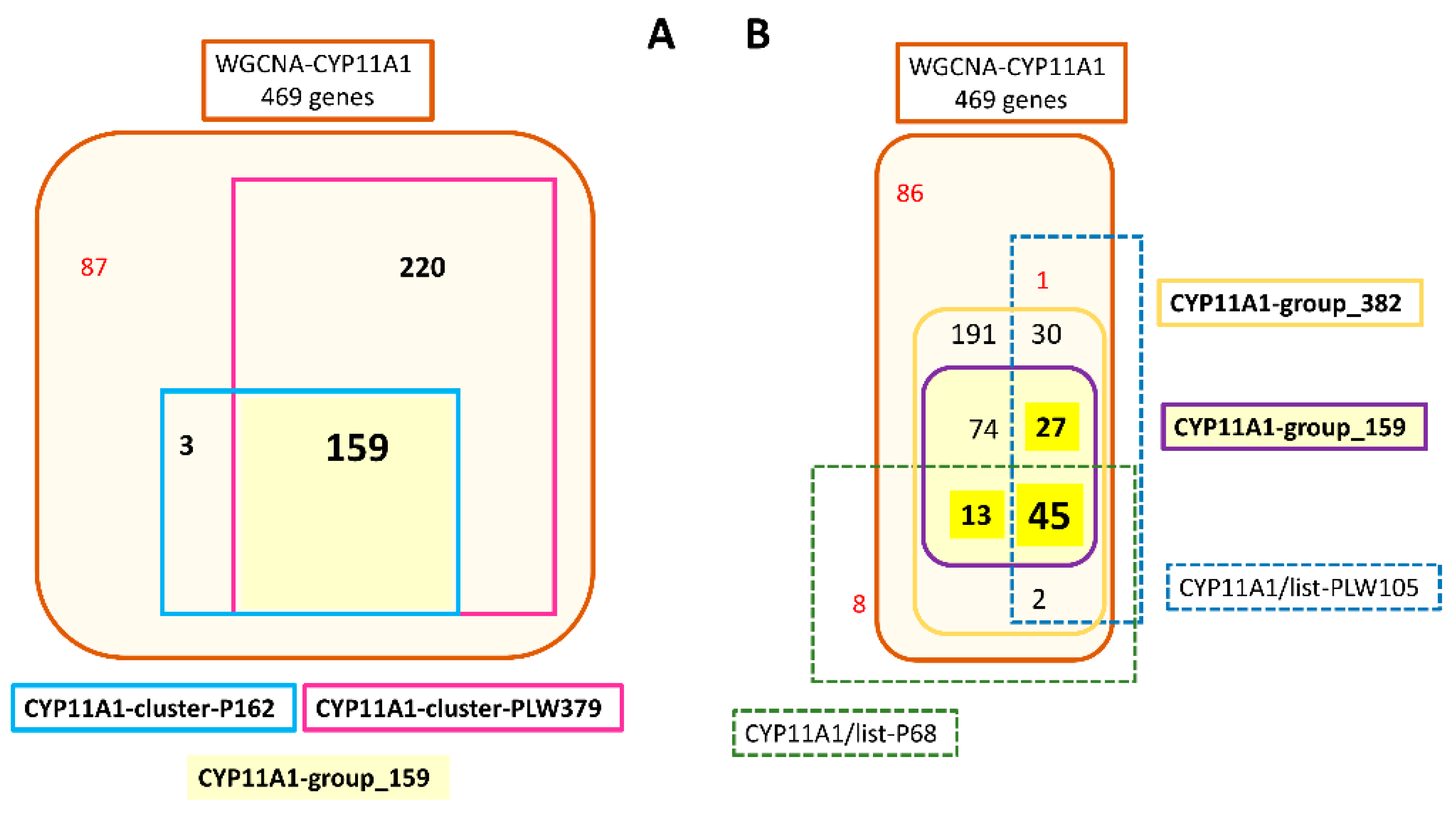
3.2.2. Identification of Correlation Modules Containing CYP11A1 and HSD17B3
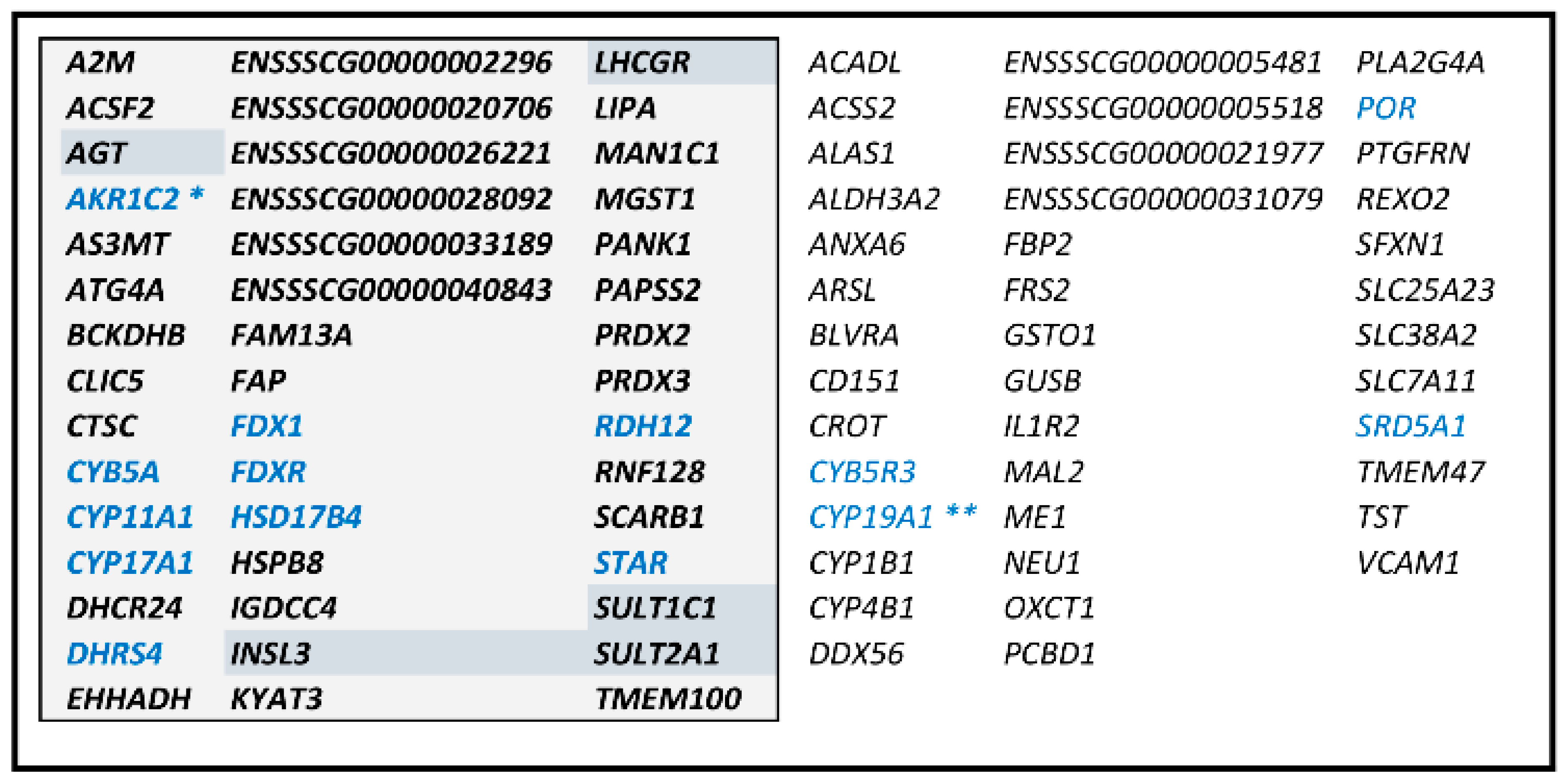
3.3. Characterization of a Correlation Group Around CYP11A1
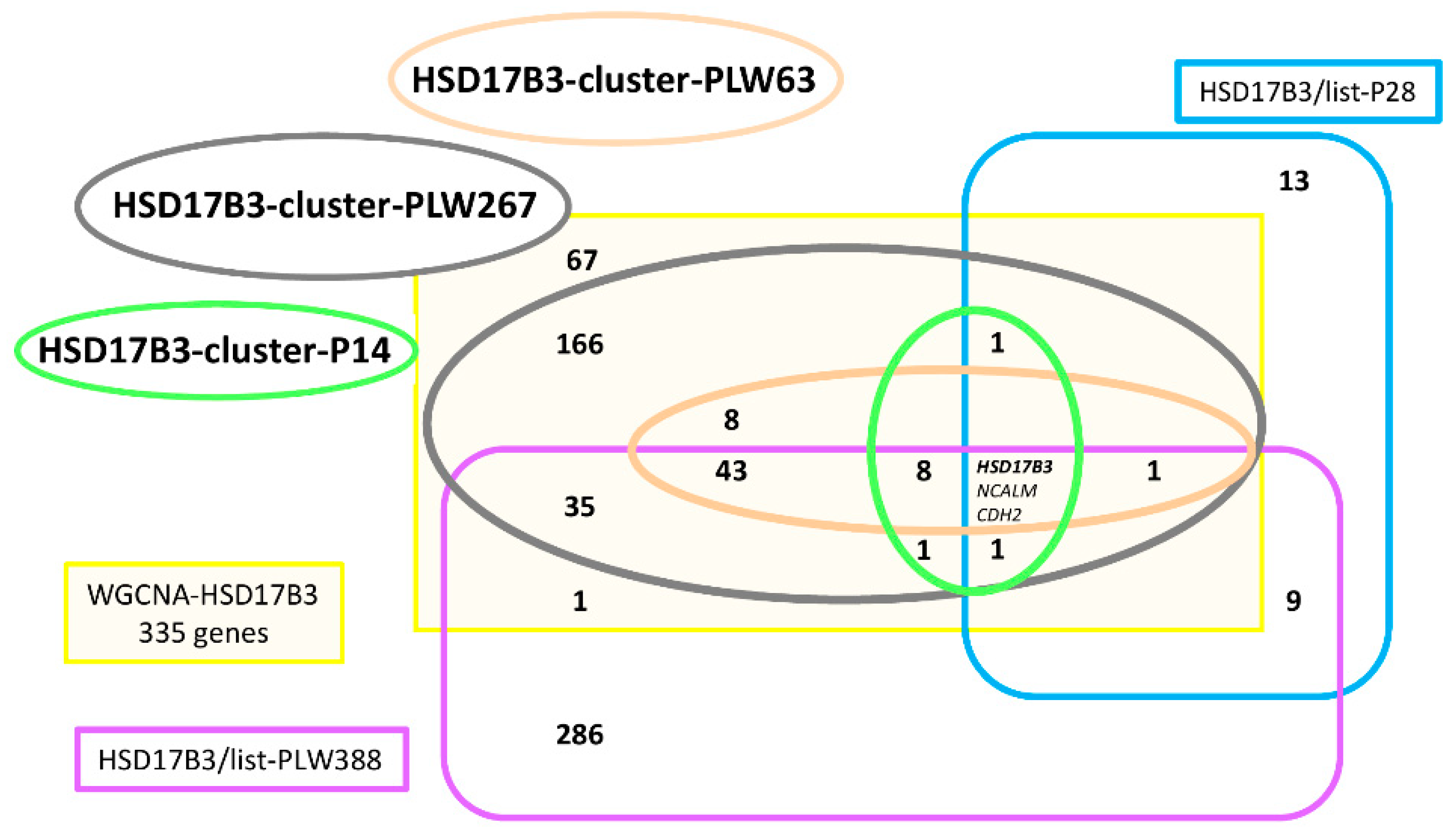
3.4. Analyses of Correlation Group around HSD17B3
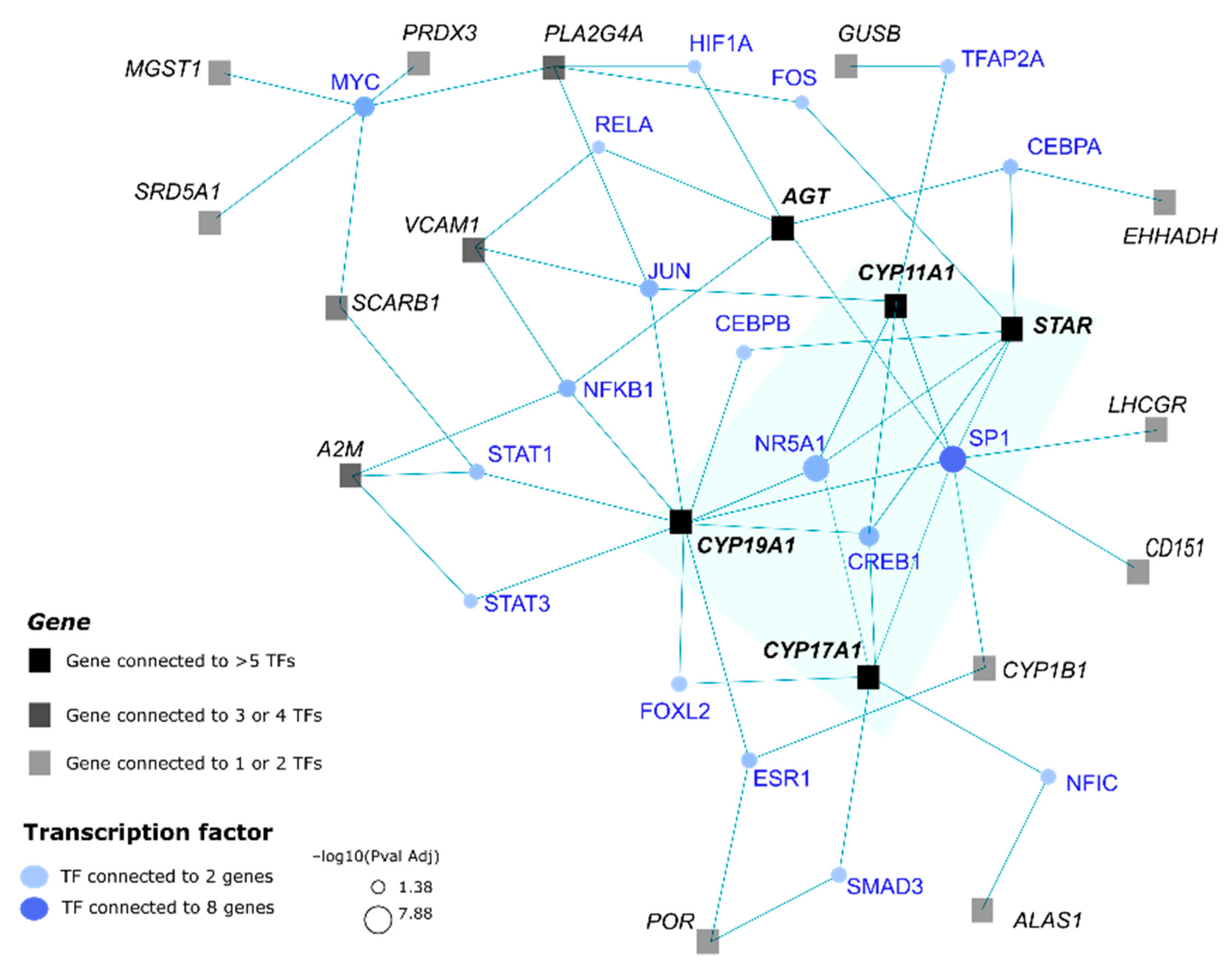
3.5. Analysis of Possible Interactions of the Genes of CYP11A1-Group by Transcriptions Factors
3.6. GNRHR2—Preliminary Results on Mature Testis
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Squires, E.J.; Bone, C.; Cameron, J. Pork Production with Entire Males: Directions for Control of Boar Taint. Animals 2020, 10, 1665. [Google Scholar] [CrossRef]
- Ubuka, T.; Son, Y.L.; Tobari, Y.; Narihiro, M.; Bentley, G.E.; Kriegsfeld, L.J.; Tsutsui, K. Central and direct regulation of testicular activity by gonadotropin-inhibitory hormone and its receptor. Front. Endocrinol. 2014, 5, 8. [Google Scholar] [CrossRef] [Green Version]
- Desaulniers, A.T.; Cederberg, R.A.; Lents, C.A.; White, B.R. Expression and Role of Gonadotropin-Releasing Hormone 2 and Its Receptor in Mammals. Front. Endocrinol. 2017, 8, 269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desaulniers, A.T.; Cederberg, R.A.; Mills, G.A.; Ford, J.J.; Lents, C.A.; White, B.R. LH-Independent Testosterone Secretion Is Mediated by the Interaction Between GNRH2 and Its Receptor Within Porcine Testes. Biol. Reprod. 2015, 93, 45. [Google Scholar] [CrossRef]
- Robic, A.; Faraut, T.; Prunier, A. Pathways and genes involved in steroid hormone metabolism in male pigs: A review and update. J. Steroid Biochem. Mol. Biol. 2014, 140, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Zirkin, B.R.; Papadopoulos, V. Leydig cells: Formation, function, and regulation. Biol. Reprod. 2018, 99, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Choi, I.; Kim, J.Y.; Lee, E.J.; Kim, Y.Y.; Chung, C.S.; Chang, J.; Choi, N.J.; Chung, H.J.; Lee, K.H. Ontogeny of expression and localization of steroidogenic enzymes in the neonatal and prepubertal pig testes. J. Androl. 2009, 30, 57–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, A.; Kaufman, J.M.; Goemaere, S.; van Pottelberg, I. Estradiol in elderly men. Aging Male Off. J. Int. Soc. Study Aging Male 2002, 5, 98–102. [Google Scholar] [CrossRef]
- Raeside, J.I.; Christie, H.L.; Renaud, R.L.; Sinclair, P.A. The boar testis: The most versatile steroid producing organ known. Soc. Reprod. Fertil. Suppl. 2006, 62, 85–97. [Google Scholar]
- Peyrat, J.P.; Meusy-Dessolle, N.; Garnier, J. Changes in Leydig cells and luteinizing hormone receptors in porcine testis during postnatal development. Endocrinology 1981, 108, 625–631. [Google Scholar] [CrossRef]
- Payne, A.H.; Hales, D.B. Overview of steroidogenic enzymes in the pathway from cholesterol to active steroid hormones. Endocr. Rev. 2004, 25, 947–970. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, F.; Ye, L.; Zirkin, B.; Chen, H. Steroidogenesis in Leydig cells: Effects of aging and environmental factors. Reproduction 2017, 154, R111–R122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graddy, L.G.; Kowalski, A.A.; Simmen, F.A.; Davis, S.L.; Baumgartner, W.W.; Simmen, R.C. Multiple isoforms of porcine aromatase are encoded by three distinct genes. J. Steroid Biochem. Mol. Biol. 2000, 73, 49–57. [Google Scholar] [CrossRef]
- Chen, M.; Yang, W.; Liu, N.; Zhang, X.; Dong, W.; Lan, X.; Pan, C. Pig Hsd17b3: Alternative splice variants expression, insertion/deletion (indel) in promoter region and their associations with male reproductive traits. J. Steroid Biochem. Mol. Biol. 2019, 195, 105483. [Google Scholar] [CrossRef]
- Penning, T.M.; Burczynski, M.E.; Jez, J.M.; Hung, C.F.; Lin, H.K.; Ma, H.; Moore, M.; Palackal, N.; Ratnam, K. Human 3alpha-hydroxysteroid dehydrogenase isoforms (AKR1C1-AKR1C4) of the aldo-keto reductase superfamily: Functional plasticity and tissue distribution reveals roles in the inactivation and formation of male and female sex hormones. Biochem. J. 2000, 351, 67–77. [Google Scholar]
- Yazawa, T.; Imamichi, Y.; Uwada, J.; Sekiguchi, T.; Mikami, D.; Kitano, T.; Ida, T.; Sato, T.; Nemoto, T.; Nagata, S.; et al. Evaluation of 17beta-hydroxysteroid dehydrogenase activity using androgen receptor-mediated transactivation. J. Steroid Biochem. Mol. Biol. 2020, 196, 105493. [Google Scholar] [CrossRef]
- Robic, A.; Feve, K.; Louveau, I.; Riquet, J.; Prunier, A. Exploration of steroidogenesis-related genes in testes, ovaries, adrenals, liver and adipose tissue in pigs. Anim. Sci. J. 2016, 87, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Robic, A.; Feve, K.; Riquet, J.; Prunier, A. Transcript levels of genes implicated in steroidogenesis in the testes and fat tissue in relation to androstenone accumulation in fat of pubertal pigs. Domest. Anim. Endocrinol. 2016, 57, 1–9. [Google Scholar] [CrossRef]
- Parois, S.P.; Prunier, A.; Mercat, M.J.; Merlot, E.; Larzul, C. Genetic relationships between measures of sexual development, boar taint, health, and aggressiveness in pigs. J. Anim. Sci. 2015, 93, 3749–3758. [Google Scholar] [CrossRef]
- Robic, A.; Faraut, T.; Djebali, S.; Weikard, R.; Feve, K.; Maman, S.; Kuehn, C. Analysis of pig transcriptomes suggests a global regulation mechanism enabling temporary bursts of circular RNAs. Rna Biol. 2019, 16, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ensembl-Pig. Porcine Genome Database. Available online: http://www.ensembl.org/Sus_scrofa/Info/Index (accessed on 4 March 2021).
- Lervik, S.; Kristoffersen, A.B.; Conley, L.N.; Oskam, I.C.; Hedegaard, J.; Ropstad, E.; Olsaker, I. Gene expression during testis development in Duroc boars. Animal 2015, 9, 1832–1842. [Google Scholar] [CrossRef] [Green Version]
- Grindflek, E.; Berget, I.; Moe, M.; Oeth, P.; Lien, S. Transcript profiling of candidate genes in testis of pigs exhibiting large differences in androstenone levels. BMC Genet 2010, 11, 4. [Google Scholar] [CrossRef] [Green Version]
- Leung, M.C.K.; Bowley, K.L.; Squires, E.J. examination of testicular Gene expression patterns in Yorkshire pigs with high and low levels of boar taint. Anim. Biotechnol. 2010, 21, 77–87. [Google Scholar] [CrossRef] [PubMed]
- R-Software. Available online: http://www.r-project.org/ (accessed on 4 March 2021).
- Grech, V. WASP (Write a Scientific Paper) using Excel—13: Correlation and Regression. Early Hum. Dev. 2018, 122, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- WGCNA-Recommandations. Available online: https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/Tutorials/ (accessed on 4 March 2021).
- SIGENAE. Available online: http://www.sigenae.org/ (accessed on 4 March 2021).
- HCA-Galaxy-Tutorial. Available online: http://genoweb.toulouse.inra.fr/~formation/CATIBIOS4BIOL_stats/Learning_clustering_current.pdf (accessed on 4 March 2021).
- Genecodis4-Software. Available online: https://genecodis.genyo.es/ (accessed on 4 March 2021).
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate—A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Garcia-Alonso, L.; Holland, C.H.; Ibrahim, M.M.; Turei, D.; Saez-Rodriguez, J. Benchmark and integration of resources for the estimation of human transcription factor activities. Genome Res. 2019, 29, 1363–1375. [Google Scholar] [CrossRef] [Green Version]
- Ingram, D.L.; Dauncey, M.J. Circadian rhythms in the pig. Comp. Biochem. Physiol. Comp. Physiol. 1985, 82, 1–5. [Google Scholar] [CrossRef]
- Prunier, A.; Brillouet, A.; Merlot, E.; Meunier-Salaun, M.C.; Tallet, C. Influence of housing and season on pubertal development, boar taint compounds and skin lesions of male pigs. Animal 2013, 7, 2035–2043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Sui, J.; Mao, C.; Li, X.; Chen, X.; Liang, C.; Wang, X.; Wang, S.H.; Jia, C. Identification of Key Pathways and Genes Related to the Development of Hair Follicle Cycle in Cashmere Goats. Genes 2021, 12, 180. [Google Scholar] [CrossRef]
- Ponsuksili, S.; Siengdee, P.; Du, Y.; Trakooljul, N.; Murani, E.; Schwerin, M.; Wimmers, K. Identification of common regulators of genes in co-expression networks affecting muscle and meat properties. PLoS ONE 2015, 10, e0123678. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Li, X.; Li, L.; Chen, H.; Ge, R.S. Insights into the Development of the Adult Leydig Cell Lineage from Stem Leydig Cells. Front. Physiol. 2017, 8, 430. [Google Scholar] [CrossRef] [Green Version]
- Ivell, R.; Alhujaili, W.; Kohsaka, T.; Anand-Ivell, R. Physiology and evolution of the INSL3/RXFP2 hormone/receptor system in higher vertebrates. Gen. Comp. Endocrinol. 2020, 299, 113583. [Google Scholar] [CrossRef]
- Soffientini, U.; Rebourcet, D.; Abel, M.H.; Lee, S.; Hamilton, G.; Fowler, P.A.; Smith, L.B.; O’Shaughnessy, P.J. Identification of Sertoli cell-specific transcripts in the mouse testis and the role of FSH and androgen in the control of Sertoli cell activity. BMC Genom. 2017, 18, 972. [Google Scholar] [CrossRef]
- Gegenschatz-Schmid, K.; Verkauskas, G.; Demougin, P.; Bilius, V.; Dasevicius, D.; Stadler, M.B.; Hadziselimovic, F. Curative GnRHa treatment has an unexpected repressive effect on Sertoli cell specific genes. Basic Clin. Androl. 2018, 28, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, C.; Stevant, I.; Borel, C.; Conne, B.; Pitetti, J.L.; Calvel, P.; Kaessmann, H.; Jegou, B.; Chalmel, F.; Nef, S. Research resource: The dynamic transcriptional profile of sertoli cells during the progression of spermatogenesis. Mol. Endocrinol. 2015, 29, 627–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, J.; Nie, X.; Giebler, M.; Mlcochova, H.; Wang, Y.; Grow, E.J.; Kim, R.; Tharmalingam, M.; Matilionyte, G.; Lindskog, C.; et al. The Dynamic Transcriptional Cell Atlas of Testis Development during Human Puberty. Cell Stem Cell 2020, 26, 262–276.e4. [Google Scholar] [CrossRef] [Green Version]
- Schuler, G.; Dezhkam, Y.; Bingsohn, L.; Hoffmann, B.; Failing, K.; Galuska, C.E.; Hartmann, M.F.; Sanchez-Guijo, A.; Wudy, S.A. Free and sulfated steroids secretion in postpubertal boars (Sus scrofa domestica). Reproduction 2014, 148, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Zimmer, B.; Tenbusch, L.; Klymiuk, M.C.; Dezhkam, Y.; Schuler, G. Sulfation pathways: Expression of SULT2A1, SULT2B1 and HSD3B1 in the porcine testis and epididymis. J. Mol. Endocrinol. 2018, 61, M41–M55. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Bennien, J.; Wapelhorst, B.; Bakhaus, K.; Schumacher, V.; Kliesch, S.; Weidner, W.; Bergmann, M.; Geyer, J.; Fietz, D. Current insights into the sulfatase pathway in human testis and cultured Sertoli cells. Histochem. Cell Biol. 2016, 146, 737–748. [Google Scholar] [CrossRef]
- Schuler, G.; Dezhkam, Y.; Tenbusch, L.; Klymiuk, M.C.; Zimmer, B.; Hoffmann, B. Sulfation pathways: Formation and hydrolysis of sulfonated estrogens in the porcine testis and epididymis. J. Mol. Endocrinol. 2018, 61, M13–M25. [Google Scholar] [CrossRef] [PubMed]
- Rebourcet, D.; Mackay, R.; Darbey, A.; Curley, M.K.; Jorgensen, A.; Frederiksen, H.; Mitchell, R.T.; O’Shaughnessy, P.J.; Nef, S.; Smith, L.B. Ablation of the canonical testosterone production pathway via knockout of the steroidogenic enzyme HSD17B3, reveals a novel mechanism of testicular testosterone production. FASEB J. 2020, 34, 10373–10386. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Claus, R. Aromatase and 11beta-hydroxysteroid dehydrogenase 2 localisation in the testes of pigs from birth to puberty linked to changes of hormone pattern and testicular morphology. Reprod. Fertil. Dev. 2008, 20, 505–512. [Google Scholar] [CrossRef]
- Schwarzenberger, F.; Toole, G.S.; Christie, H.L.; Raeside, J.I. Plasma levels of several androgens and estrogens from birth to puberty in male domestic pigs. Acta Endocrinol. 1993, 128, 173–177. [Google Scholar] [CrossRef]
- Michaelis, M.; Sobczak, A.; Koczan, D.; Langhammer, M.; Reinsch, N.; Schoen, J.; Weitzel, J.M. Selection for female traits of high fertility affects male reproductive performance and alters the testicular transcriptional profile. BMC Genom. 2017, 18, 889. [Google Scholar] [CrossRef] [Green Version]
- Minagawa, I.; Sagata, D.; Pitia, A.M.; Kohriki, H.; Shibata, M.; Sasada, H.; Hasegawa, Y.; Kohsaka, T. Dynamics of insulin-like factor 3 and its receptor expression in boar testes. J. Endocrinol. 2014, 220, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Facondo, P.; Delbarba, A.; Maffezzoni, F.; Cappelli, C.; Ferlin, A. INSL3: A Marker of Leydig Cell Function and Testis-Bone-Skeletal Muscle Network. Protein Pept. Lett. 2020, 27, 1246–1252. [Google Scholar] [CrossRef]
- Minagawa, I.; Murata, Y.; Terada, K.; Shibata, M.; Park, E.Y.; Sasada, H.; Kohsaka, T. Evidence for the role of INSL3 on sperm production in boars by passive immunisation. Andrologia 2018, 50, e13010. [Google Scholar] [CrossRef]
- Ding, H.; Liu, M.; Zhou, C.; You, X.; Suo, Z.; Zhang, C.; Xu, D. Expression and regulation of GnRHR2 gene and testosterone secretion mediated by GnRH2 and GnRHR2 within porcine testes. J. Steroid Biochem. Mol. Biol. 2019, 190, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Desaulniers, A.T.; Cederberg, R.A.; Mills, G.A.; Lents, C.A.; White, B.R. Production of a gonadotropin-releasing hormone 2 receptor knockdown (GNRHR2 KD) swine line. Transgenic Res. 2017, 26, 567–575. [Google Scholar] [CrossRef] [Green Version]
- Bajpai, R.; Nagaraju, G.P. Specificity protein 1: Its role in colorectal cancer progression and metastasis. Crit. Rev. Oncol./Hematol. 2017, 113, 1–7. [Google Scholar] [CrossRef]
- Sugawara, T.; Saito, M.; Fujimoto, S. Sp1 and SF-1 interact and cooperate in the regulation of human steroidogenic acute regulatory protein gene expression. Endocrinology 2000, 141, 2895–2903. [Google Scholar] [CrossRef] [PubMed]
- Jeyasuria, P.; Ikeda, Y.; Jamin, S.P.; Zhao, L.; De Rooij, D.G.; Themmen, A.P.; Behringer, R.R.; Parker, K.L. Cell-specific knockout of steroidogenic factor 1 reveals its essential roles in gonadal function. Mol. Endocrinol. 2004, 18, 1610–1619. [Google Scholar] [CrossRef] [Green Version]
- Convissar, S.; Winston, N.J.; Fierro, M.A.; Scoccia, H.; Zamah, A.M.; Stocco, C. Sp1 regulates steroidogenic genes and LHCGR expression in primary human luteinized granulosa cells. J. Steroid Biochem. Mol. Biol. 2019, 190, 183–192. [Google Scholar] [CrossRef]
- Choi, H.; Ryu, K.Y.; Roh, J. Kruppel-like factor 4 plays a role in the luteal transition in steroidogenesis by downregulating Cyp19A1 expression. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E1071–E1080. [Google Scholar] [CrossRef]
- Sirotkin, A.V.; Benco, A.; Mlyncek, M.; Harrath, A.H.; Alwasel, S.; Kotwica, J. The involvement of the phosphorylatable and nonphosphorylatable transcription factor CREB-1 in the control of human ovarian cell functions. Comptes Rendus Biol. 2019, 342, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Roumaud, P.; Rwigemera, A.; Martin, L.J. Transcription factors SF1 and cJUN cooperate to activate the Fdx1 promoter in MA-10 Leydig cells. J. Steroid Biochem. Mol. Biol. 2017, 171, 121–132. [Google Scholar] [CrossRef]
- Tremblay, M.A.; Mendoza-Villarroel, R.E.; Robert, N.M.; Bergeron, F.; Tremblay, J.J. KLF6 cooperates with NUR77 and SF1 to activate the human INSL3 promoter in mouse MA-10 leydig cells. J. Mol. Endocrinol. 2016, 56, 163–173. [Google Scholar] [CrossRef] [Green Version]
- Brauer, V.M.; Wiarda-Bell, J.R.; Desaulniers, A.T.; Cederberg, R.A.; White, B.R. Functional activity of the porcine Gnrhr2 gene promoter in testis-derived cells is partially conferred by nuclear factor-kappaB, specificity protein 1 and 3 (SP1/3) and overlapping early growth response 1/SP1/3 binding sites. Gene 2016, 587, 137–146. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Robic, A.; Faraut, T.; Feve, K.; Djebali, S.; Prunier, A.; Larzul, C.; Liaubet, L. Correlation Networks Provide New Insights into the Architecture of Testicular Steroid Pathways in Pigs. Genes 2021, 12, 551. https://doi.org/10.3390/genes12040551
Robic A, Faraut T, Feve K, Djebali S, Prunier A, Larzul C, Liaubet L. Correlation Networks Provide New Insights into the Architecture of Testicular Steroid Pathways in Pigs. Genes. 2021; 12(4):551. https://doi.org/10.3390/genes12040551
Chicago/Turabian StyleRobic, Annie, Thomas Faraut, Katia Feve, Sarah Djebali, Armelle Prunier, Catherine Larzul, and Laurence Liaubet. 2021. "Correlation Networks Provide New Insights into the Architecture of Testicular Steroid Pathways in Pigs" Genes 12, no. 4: 551. https://doi.org/10.3390/genes12040551
APA StyleRobic, A., Faraut, T., Feve, K., Djebali, S., Prunier, A., Larzul, C., & Liaubet, L. (2021). Correlation Networks Provide New Insights into the Architecture of Testicular Steroid Pathways in Pigs. Genes, 12(4), 551. https://doi.org/10.3390/genes12040551