
A New Pathogenic Variant in POU3F4 Causing Deafness Due to an Incomplete Partition of the Cochlea Paved the Way for Innovative Surgery

 ,
,
Abstract
:1. Introduction
1.1. POU3F4 Gene
1.2. Molecular and Clinical Characterization of POU3F4 Pathogenic Variants (Incl. Surgical Challenges)
1.3. Cochlear Implantation Surgery for Sensorineural Hearing Loss
2. Materials and Methods
2.1. The Patient
2.2. Audiological Evaluation
2.3. Molecular Analysis
2.4. RACIS: Robotically-Assisted Cochlear Implant Surgery
3. Results
3.1. Surgery Results
3.2. Molecular Genetic Analysis
3.3. Audiological Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Morton, N. Genetic epidemiology of hearing impairment. Ann. N. Y. Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef]
- Morton, C.C.; Nance, W.E. Newborn hearing screening—a silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Sennaroğlu, L.; Bajin, M.D. Classification and current management of inner ear malformations. Balk. Med. J. 2017, 34, 397. [Google Scholar] [CrossRef] [PubMed]
- De Kok, Y.; van der Maarel, S.M.; Bitner-Glindzicz, M.; Huber, I.; Monaco, A.P.; Malcolm, S.; Pembrey, M.E.; Ropers, H.-H.; Cremers, F. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 1995, 267, 685–688. [Google Scholar] [CrossRef] [Green Version]
- de Kok, Y.J.; Vossenaar, E.R.; Cremers, C.W.; Dahl, N.; Laporte, J.; Jia Hu, L.; Lacombe, D.; Fischel-Ghodsian, N.; Friedman, R.A.; Parnes, L.S.; et al. Identification of a hot spot for microdeletions in patients with X-linked deafness type 3 (DFN3) 900 kb proximal to the DFN3 gene POU3F4. Hum. Mol. Genet. 1996, 5, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Kanno, A.; Mutai, H.; Namba, K.; Morita, N.; Nakano, A.; Ogahara, N.; Sugiuchi, T.; Ogawa, K.; Matsunaga, T. Frequency and specific characteristics of the incomplete partition type III anomaly in children. Laryngoscope 2017, 127, 1663–1669. [Google Scholar] [CrossRef]
- Douville, P.; Atanasoski, S.; Tobler, A.; Fontana, A.; Schwab, M. The brain-specific POU-box gene Brn4 is a sex-linked transcription factor located on the human and mouse X chromosomes. Mamm. Genome 1994, 5, 180–182. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, D.H.; Chung, T.; Chang, M.; Kim, E.H.; Kim, A.R.; Seok, J.; Chang, S.O.; Bok, J.; Kim, D.; et al. Destabilization and Mislocalization of POU 3 F 4 by C-Terminal Frameshift Truncation and Extension Mutation. Hum. Mutat. 2013, 34, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Mathis, J.M.; Simmons, D.; He, X.; Swanson, L.; Rosenfeld, M. Brain 4: A novel mammalian POU domain transcription factor exhibiting restricted brain-specific expression. EMBO J. 1992, 11, 2551–2561. [Google Scholar] [CrossRef]
- Kok, Y.J.d.; Cremers, C.W.; Ropers, H.H.; Cremers, F.P. The molecular basis of X-linked deafness type 3 (DFN3) in two sporadic cases: Identification of a somatic mosaicism for a POU3F4 missense mutation. Hum. Mutat. 1997, 10, 207–211. [Google Scholar] [CrossRef]
- Brooks, P.M.; Rose, K.P.; MacRae, M.L.; Rangoussis, K.M.; Gurjar, M.; Hertzano, R.; Coate, T.M. Pou3f4-expressing otic mesenchyme cells promote spiral ganglion neuron survival in the postnatal mouse cochlea. J. Comp. Neurol. 2020, 528, 1967–1985. [Google Scholar] [CrossRef]
- Samadi, D.S.; Saunders, J.C.; Crenshaw, E.B., III. Mutation of the POU-domain gene Brn4/Pou3f4 affects middle-ear sound conduction in the mouse. Hear. Res. 2005, 199, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Song, M.H.; Choi, S.-Y.; Wu, L.; Oh, S.-K.; Lee, H.K.; Lee, D.J.; Shim, D.-B.; Choi, J.Y.; Kim, U.-K.; Bok, J. Pou3f4 deficiency causes defects in otic fibrocytes and stria vascularis by different mechanisms. Biochem. Biophys. Res. Commun. 2011, 404, 528–533. [Google Scholar] [CrossRef]
- Choi, B.Y.; An, Y.H.; Song, J.J.; Koo, J.W.; Lee, J.H.; Oh, S.H.; Chang, S.O.; Kim, C.S.; Park, J.H. Clinical observations and molecular variables of patients with hearing loss and incomplete partition type III. Laryngoscope 2016, 126, E123–E128. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Song, M.H.; Kang, M.; Lee, J.T.; Kong, K.-A.; Choi, S.-J.; Lee, K.Y.; Venselaar, H.; Vriend, G.; Lee, W.-S.; et al. Clinical and molecular characterizations of novel POU3F4 mutations reveal that DFN3 is due to null function of POU3F4 protein. Physiol. Genom. 2009, 39, 195–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corvino, V.; Apisa, P.; Malesci, R.; Laria, C.; Auletta, G.; Franzé, A. X-linked sensorineural hearing loss: A literature review. Curr. Genom. 2018, 19, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Bitner-Glindzicz, M.; Turnpenny, P.; Höglund, P.; Kääriäinen, H.; Sankila, E.-M.; van der Maarel, S.M.; de Kok, Y.J.; Ropers, H.-H.; Cremers, F.P.; Pembrey, M. Further mutations in Brain 4 (POU3F4) clarify the phenotype in the X-linked deafness, DFN3. Hum. Mol. Genet. 1995, 4, 1467–1469. [Google Scholar] [CrossRef] [Green Version]
- Vore, A.P.; Chang, E.H.; Hoppe, J.E.; Butler, M.G.; Forrester, S.; Schneider, M.C.; Smith, L.L.; Burke, D.W.; Campbell, C.A.; Smith, R.J. Deletion of and novel missense mutation in POU3F4 in 2 families segregating X-linked nonsyndromic deafness. Arch. Otolaryngol. Head Neck Surg. 2005, 131, 1057–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.J.; Li, Q.Z.; Rao, S.Q.; Zhao, Y.L.; Yuan, H.; Yang, W.Y.; Han, D.y.; Shen, Y. A novel mutation of POU3F4 causes congenital profound sensorineural hearing loss in a large Chinese family. Laryngoscope 2006, 116, 944–950. [Google Scholar] [CrossRef]
- Li, J.; Cheng, J.; Lu, Y.; Lu, Y.; Chen, A.; Sun, Y.; Kang, D.; Zhang, X.; Dai, P.; Han, D. Identification of a novel mutation in POU3F4 for prenatal diagnosis in a Chinese family with X-linked nonsyndromic hearing loss. J. Genet. Genom. 2010, 37, 787–793. [Google Scholar] [CrossRef]
- Waryah, A.M.; Ahmed, Z.M.; Binder, M.A.; Choo, D.I.; Sisk, R.A.; Shahzad, M.; Khan, S.N.; Friedman, T.B.; Riazuddin, S.; Riazuddin, S. Molecular and clinical studies of X-linked deafness among Pakistani families. J. Hum. Genet. 2011, 56, 534–540. [Google Scholar] [CrossRef]
- Bademci, G.; Lasisi, A.O.; Yariz, K.O.; Montenegro, P.; Menendez, I.; Vinueza, R.; Paredes, R.; Moreta, G.; Subasioglu, A.; Blanton, S. Novel domain-specific POU3F4 mutations are associated with X-linked deafness: Examples from different populations. Bmc Med. Genet. 2015, 16, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Barashkov, N.A.; Klarov, L.A.; Teryutin, F.M.; Solovyev, A.V.; Pshennikova, V.G.; Konnikova, E.E.; Romanov, G.P.; Tobokhov, A.V.; Morozov, I.V.; Bondar, A.A.; et al. A novel pathogenic variant c. 975G > A (p. Trp325*) in the POU3F4 gene in Yakut family (Eastern Siberia, Russia) with the X-linked deafness-2 (DFNX2). Int. J. Pediatr. Otorhinolaryngol. 2018, 104, 94–97. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.-M.; Jie, H.-Q.; Wang, H.; Wu, Y.-Q.; Chen, Z.-N.; Xing, Y.-Z.; Wang, J.-P.; Shi, H.-B.; Yin, S.-K. A novel POU domain class 3 transcription factor 4 mutation causes X-linked non-syndromic hearing loss in a Chinese family. Chin. Med. J. 2019, 132, 2251–2253. [Google Scholar] [CrossRef]
- Han, J.J.; Nguyen, P.D.; Oh, D.-Y.; Han, J.H.; Kim, A.-R.; Kim, M.Y.; Park, H.-R.; Tran, L.H.; Dung, N.H.; Koo, J.-W. Elucidation of the unique mutation spectrum of severe hearing loss in a Vietnamese pediatric population. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Hagiwara, H.; Tamagawa, Y.; Kitamura, K.; Kodera, K. A new mutation in the POU3F4 gene in a japanese family with x-linked mixed deafness (DFN3). Laryngoscope 1998, 108, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Moteki, H.; Shearer, A.E.; Izumi, S.; Kubota, Y.; Azaiez, H.; Booth, K.T.; Sloan, C.M.; Kolbe, D.L.; Smith, R.J.; Usami, S.-i. De Novo Mutation in X-Linked Hearing Loss–Associated POU3F4 in a Sporadic Case of Congenital Hearing Loss. Ann. Otol. Rhinol. Laryngol. 2015, 124, 169S–176S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marlin, S.; Moizard, M.; David, A.; Chaissang, N.; Raynaud, M.; Jonard, L.; Feldmann, D.; Loundon, N.; Denoyelle, F.; Toutain, A. Phenotype and genotype in females with POU3F4 mutations. Clin. Genet. 2009, 76, 558–563. [Google Scholar] [CrossRef]
- Chao, X.; Xiao, Y.; Zhang, F.; Luo, J.; Wang, R.; Liu, W.; Wang, H.; Xu, L. Cochlear Implantation in a Patient with a Novel POU3F4 Mutation and Incomplete Partition Type-III Malformation. Neural Plast. 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Giannantonio, S.; Agolini, E.; Scorpecci, A.; Anzivino, R.; Bellacchio, E.; Cocciadiferro, D.; Novelli, A.; Digilio, M.C.; Marsella, P. Genetic identification and molecular modeling characterization of a novel POU3F4 variant in two Italian deaf brothers. Int. J. Pediatr. Otorhinolaryngol. 2020, 129, 109790. [Google Scholar] [CrossRef] [PubMed]
- Bykhovskaya, Y.; Wilson, D.F.; Friedman, R.A.; Tu, G.; Farnes, L.S.; Talbot, J.M.; Fischel-Ghodsian, N. Molecular analysis of the POU3F4 gene in patients with clinical and radiographic evidence of X-linked mixed deafness with perilymphatic gusher. Ann. Otol. Rhinol. Laryngol. 1997, 106, 320–325. [Google Scholar] [CrossRef]
- Arellano, B.; Camacho, R.R.; Berrocal, J.R.G.; Villamar, M.; del Castillo, I.; Moreno, F. Sensorineural hearing loss and Mondini dysplasia caused by a deletion at locus DFN3. Arch. Otolaryngol. Head Neck Surg. 2000, 126, 1065–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.K.; Lee, S.H.; Lee, K.Y.; Lim, E.J.; Choi, S.Y.; Park, R.K.; Kim, U.K. Novel POU3F4 mutations and clinical features of DFN3 patients with cochlear implants. Clin. Genet. 2009, 75, 572–575. [Google Scholar] [PubMed]
- Stankovic, K.M.; Hennessey, A.M.; Herrmann, B.; Mankarious, L.A. Cochlear implantation in children with congenital X-linked deafness due to novel mutations in POU3F4 gene. Ann. Otol. Rhinol. Laryngol. 2010, 119, 815–822. [Google Scholar] [CrossRef]
- Schild, C.; Prera, E.; Lüblinghoff, N.; Arndt, S.; Aschendorff, A.; Birkenhäger, R. Novel mutation in the homeobox domain of transcription factor POU3F4 associated with profound sensorineural hearing loss. Otol. Neurotol. 2011, 32, 690–694. [Google Scholar] [CrossRef] [PubMed]
- Parzefall, T.; Shivatzki, S.; Lenz, D.R.; Rathkolb, B.; Ushakov, K.; Karfunkel, D.; Shapira, Y.; Wolf, M.; Mohr, M.; Wolf, E.; et al. Cytoplasmic mislocalization of POU3F4 due to novel mutations leads to deafness in humans and mice. Hum. Mutat. 2013, 34, 1102–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollak, A.; Lechowicz, U.; Kędra, A.; Stawiński, P.; Rydzanicz, M.; Furmanek, M.; Brzozowska, M.; Mrówka, M.; Skarżyński, H.; Skarżyński, P.H. Novel and de novo mutations extend association of POU3F4 with distinct clinical and radiological phenotype of hearing loss. PLoS ONE 2016, 11, e0166618. [Google Scholar] [CrossRef]
- Lin, Y.-H.; Lin, Y.-H.; Lu, Y.-C.; Liu, T.-C.; Chen, C.-Y.; Hsu, C.-J.; Chen, P.-L.; Wu, C.-C. A novel missense variant in the nuclear localization signal of POU4F3 causes autosomal dominant non-syndromic hearing loss. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Du, W.; Han, M.-K.; Wang, D.-Y.; Han, B.; Zong, L.; Lan, L.; Yang, J.; Shen, Q.; Xie, L.-Y.; Yu, L.; et al. A POU3F4 mutation causes nonsyndromic hearing loss in a Chinese X-linked recessive family. Chin. Med. 2017, 130, 88–92. [Google Scholar] [CrossRef]
- Su, Y.; Gao, X.; Huang, S.-S.; Mao, J.-N.; Huang, B.-Q.; Zhao, J.-D.; Kang, D.-Y.; Zhang, X.; Dai, P. Clinical and molecular characterization of POU3F4 mutations in multiple DFNX2 Chinese families. Bmc Med. Genet. 2018, 19, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Oh, J.; Han, J.H.; Park, H.-R.; Kim, B.J.; Lee, S.; Kim, M.Y.; Lee, S.; Oh, D.-Y.; Choung, Y.-H. Identification of a Novel Frameshift Variant of POU3F4 and Genetic Counseling of Korean Incomplete Partition Type III Subjects Based on Detailed Genotypes. Genet. Test. Mol. Biomark. 2019, 23, 423–427. [Google Scholar] [CrossRef]
- Wu, D.; Huang, W.; Xu, Z.; Li, S.; Zhang, J.; Chen, X.; Tang, Y.; Qiu, J.; Wang, Z.; Duan, X.; et al. Clinical and genetic study of 12 Chinese Han families with nonsyndromic deafness. Mol. Genet. Genom. Med. 2020, 8, e1177. [Google Scholar] [CrossRef] [Green Version]
- Nance, W.; Setleff, R.; McLeod, A.; Sweeney, A.; Cooper, C.; McConnell, F. X-linked mixed deafness with congenital fixation of the stapedial footplate and perilymphatic gusher. Birth Defects Orig. Artic. Ser. 1971, 7, 64–69. [Google Scholar]
- Papadaki, E.; Prassopoulos, P.; Bizakis, J.; Karampekios, S.; Papadakis, H.; Gourtsoyiannis, N. X-linked deafness with stapes gusher in females. Eur. J. Radiol. 1998, 29, 71–75. [Google Scholar] [CrossRef]
- Sennaroğlu, L.; Bajin, M.D. Incomplete partition type III: A rare and difficult cochlear implant surgical indication. Auris Nasus Larynx 2018, 45, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Phelps, P.; Reardon, W.; Pembrey, M.; Bellman, S.; Luxom, L. X-linked deafness, stapes gushers and a distinctive defect of the inner ear. Neuroradiology 1991, 33, 326–330. [Google Scholar] [CrossRef]
- Talbot, J.M.; Wilson, D.F. Computed tomographic diagnosis of X-linked congenital mixed deafness, fixation of the stapedial footplate, and perilymphatic gusher. Am. J. Otol. 1994, 15, 177–182. [Google Scholar] [PubMed]
- Jackler, R.K.; Luxfor, W.M.; House, W.F. Congenital malformations of the inner ear: A classification based on embryogenesis. Laryngoscope 1987, 97, 2–14. [Google Scholar] [CrossRef]
- Sennaroglu, L.; Saatci, I. A new classification for cochleovestibular malformations. Laryngoscope 2002, 112, 2230–2241. [Google Scholar] [CrossRef]
- Sennaroglu, L.; Sarac, S.; Ergin, T. Surgical results of cochlear implantation in malformed cochlea. Otol. Neurotol. 2006, 27, 615–623. [Google Scholar] [CrossRef]
- Gong, W.-X.; Gong, R.-Z.; Zhao, B. HRCT and MRI findings in X-linked non-syndromic deafness patients with a POU3F4 mutation. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 1756–1762. [Google Scholar] [CrossRef] [PubMed]
- Dhanasingh, A.; Dietz, A.; Jolly, C.; Roland, P. Human inner-ear malformation types captured in 3D. J. Int. Adv. Otol. 2019, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Smeds, H.; Wales, J.; Asp, F.; Löfkvist, U.; Falahat, B.; Anderlid, B.-M.; Anmyr, L.; Karltorp, E. X-linked malformation and cochlear implantation. Otol. Neurotol. 2017, 38, 38–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, H.; Wang, L.; Gao, F.; Liang, W.; Peng, K. Cochlear implantation using a custom guide catheter in 14 patients with incomplete partition type III. Clin. Otolaryngol. 2018, 43, 1379–1383. [Google Scholar] [CrossRef]
- Cosetti, M.K.; Friedmann, D.R.; Heman-Ackah, S.E.; Perez, R.; Waltzman, S.B.; Roland, J.T., Jr. Surgical techniques and outcomes of cochlear implantation in patients with radiographic findings consistent with X-linked deafness. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Alballaa, A.; Aschendorff, A.; Arndt, S.; Hildenbrand, T.; Becker, C.; Hassepass, F.; Laszig, R.; Beck, R.; Speck, I.; Wesarg, T.; et al. Incomplete partition type III revisited—Long-term results following cochlear implant. HNO 2020, 68, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Sun, J. Outcomes of cochlear implantation in patients with incomplete partition type III. Int. J. Pediatr. Otorhinolaryngol. 2020, 131, 109890. [Google Scholar] [CrossRef]
- Saeed, H.; Powell, H.R.; Saeed, S.R. Cochlear implantation in X-linked deafness–How to manage the surgical challenges. Cochlear Implant. Int. 2016, 17, 178–183. [Google Scholar] [CrossRef]
- Sennaroglu, L. Cochlear implantation in inner ear malformations—A review article. Cochlear Implant. Int. 2009, 11, 4–41. [Google Scholar] [CrossRef]
- Eftekharian, A.; Amizadeh, M. Cerebrospinal fluid gusher in cochlear implantation. Cochlear Implant. Int. 2014, 15, 179–184. [Google Scholar] [CrossRef]
- Sennaroğlu, L.; Atay, G.; Bajin, M.D. A new cochlear implant electrode with a “cork”-type stopper for inner ear malformations. Auris Nasus Larynx 2014, 41, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Incesulu, A.; Adapinar, B.; Kecik, C. Cochlear implantation in cases with incomplete partition type III (X-linked anomaly). Eur. Arch. Otorhinolaryngol. 2008, 265, 1425–1430. [Google Scholar] [CrossRef]
- Topsakal, V.; Matulic, M.; Assadi, M.Z.; Mertens, G.; Van Rompaey, V.; Van de Heyning, P. Comparison of the surgical techniques and robotic techniques for cochlear implantation in terms of the trajectories toward the inner ear. J. Int. Adv. Otol. 2020, 16, 3. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Cremers, C.; Snik, A.; Huygen, P.; Joosten, F.; Cremers, F. X-linked mixed deafness syndrome with congenital fixation of the stapedial footplate and perilymphatic gusher (DFN3). Adv. Otorhinolaryngol. 2002, 61, 161–167. [Google Scholar] [PubMed]
- Snik, A.F.; Hombergen, G.C.; Mylanus, E.A.; Cremers, C.W. Air-bone gap in patients with X-linked stapes gusher syndrome. Am. J. Otol. 1995, 16, 241–246. [Google Scholar]
- Mertens, G.; Andries, E.; Claes, A.J.; Topsakal, V.; Van de Heyning, P.; Van Rompaey, V.; Calvino, M.; Cuadrado, I.S.; Muñoz, E.; Gavilán, J.; et al. Cognitive Improvement After Cochlear Implantation in Older Adults with Severe or Profound Hearing Impairment: A Prospective, Longitudinal, Controlled, Multicenter Study. Ear Hear. 2021. Advance online publication. [Google Scholar] [CrossRef]
- Mosnier, I.; Vanier, A.; Bonnard, D.; Lina-Granade, G.; Truy, E.; Bordure, P.; Godey, B.; Marx, M.; Lescanne, E.; Venail, F.; et al. Long-term cognitive prognosis of profoundly deaf older adults after hearing rehabilitation using cochlear implants. J. Am. Geriatr. Soc. 2018, 66, 1553–1561. [Google Scholar] [CrossRef]
- Wick, C.C.; Kallogjeri, D.; McJunkin, J.L.; Durakovic, N.; Holden, L.K.; Herzog, J.A.; Firszt, J.B.; Buchman, C.A.; Group, C.S. Hearing and quality-of-life outcomes after cochlear implantation in adult hearing aid users 65 years or older: A secondary analysis of a nonrandomized clinical trial. Jama Otolaryngol. Head Neck Surg. 2020, 146, 925–932. [Google Scholar] [CrossRef]
- Jackler, R.K.; Luxford, W.M.; House, W.F. Sound detection with the cochlear implant in five ears of four children with congenital malformations of the cochlea. Laryngoscope 1987, 97, 15–17. [Google Scholar] [CrossRef]
- Weber, S.; Gavaghan, K.; Wimmer, W.; Williamson, T.; Gerber, N.; Anso, J.; Bell, B.; Feldmann, A.; Rathgeb, C.; Matulic, M.B.; et al. Instrument flight to the inner ear. Sci. Robot. 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Torres, R.; Kazmitcheff, G.; Bernardeschi, D.; De Seta, D.; Bensimon, J.L.; Ferrary, E.; Sterkers, O.; Nguyen, Y. Variability of the mental representation of the cochlear anatomy during cochlear implantation. Eur. Arch. Otorhinolaryngol. 2016, 273, 2009–2018. [Google Scholar] [CrossRef] [PubMed]
- Labadie, R.F.; Balachandran, R.; Noble, J.H.; Blachon, G.S.; Mitchell, J.E.; Reda, F.A.; Dawant, B.M.; Fitzpatrick, J.M. Minimally invasive image-guided cochlear implantation surgery: First report of clinical implementation. Laryngoscope 2014, 124, 1915–1922. [Google Scholar] [CrossRef] [Green Version]
- Miroir, M.; Nguyen, Y.; Kazmitcheff, G.; Ferrary, E.; Sterkers, O.; Grayeli, A.B. Friction force measurement during cochlear implant insertion: Application to a force-controlled insertion tool design. Otol. Neurotol. 2012, 33, 1092–1100. [Google Scholar] [CrossRef]
- Caversaccio, M.; Gavaghan, K.; Wimmer, W.; Williamson, T.; Ansò, J.; Mantokoudis, G.; Gerber, N.; Rathgeb, C.; Feldmann, A.; Wagner, F.; et al. Robotic cochlear implantation: Surgical procedure and first clinical experience. Acta Otolaryngol. 2017, 137, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Ansó, J.; Dür, C.; Apelt, M.; Venail, F.; Scheidegger, O.; Seidel, K.; Rohrbach, H.; Forterre, F.; Dettmer, M.S.; Zlobec, I. Prospective validation of facial nerve monitoring to prevent nerve damage during robotic drilling. Front. Surg. 2019, 6, 58. [Google Scholar] [CrossRef]
- Wootten, C.T.; Backous, D.D.; Haynes, D.S. Management of cerebrospinal fluid leakage from cochleostomy during cochlear implant surgery. Laryngoscope 2006, 116, 2055–2059. [Google Scholar] [CrossRef]
- Kang, W.S.; Shim, B.S.; Lee, K.-S. Audiologic performance after cochlear implantation in children with X-linked deafness: Comparison with deaf children with a normal inner ear structure. Otol. Neurotol. 2013, 34, 544–548. [Google Scholar] [CrossRef]
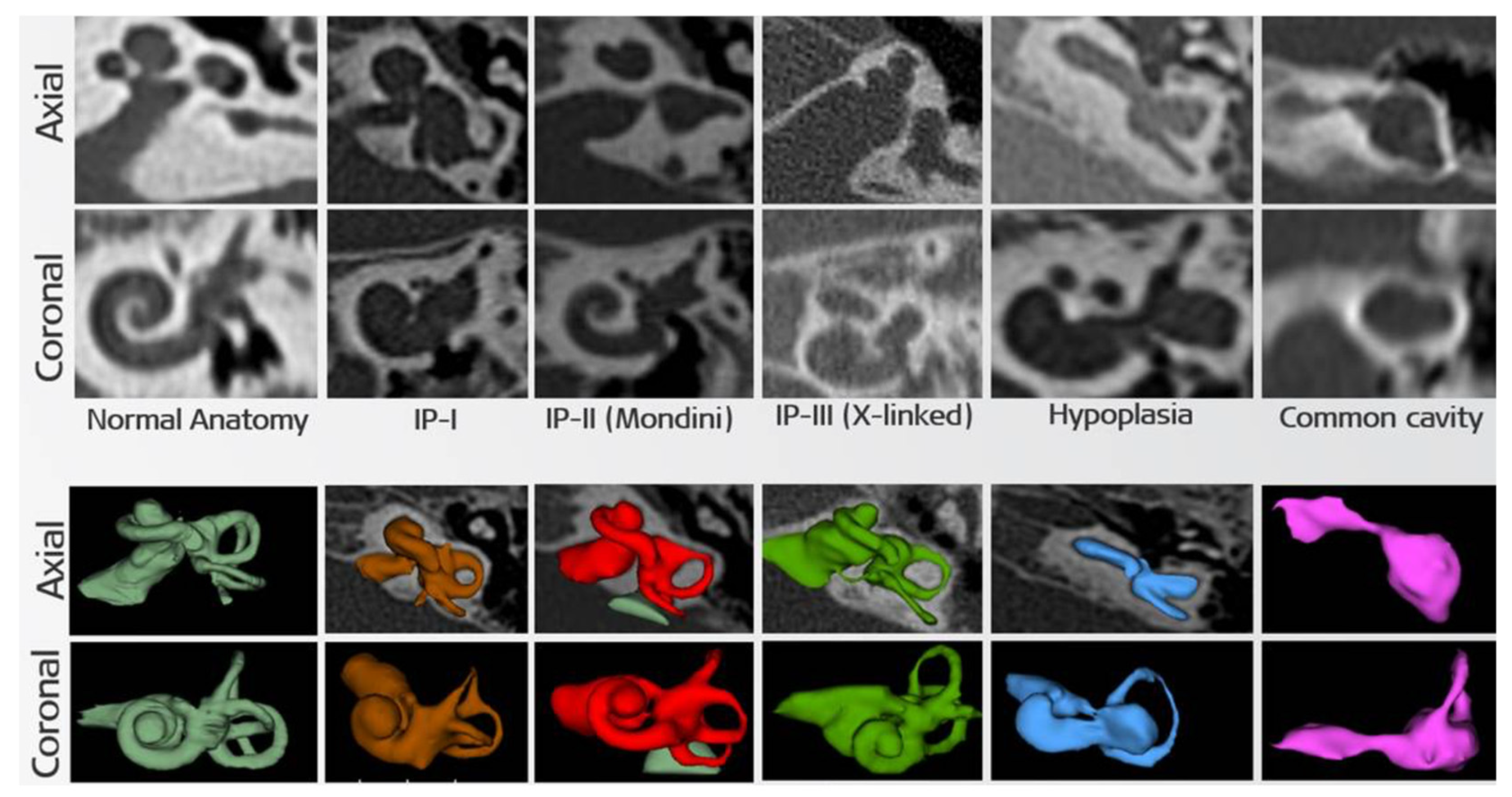
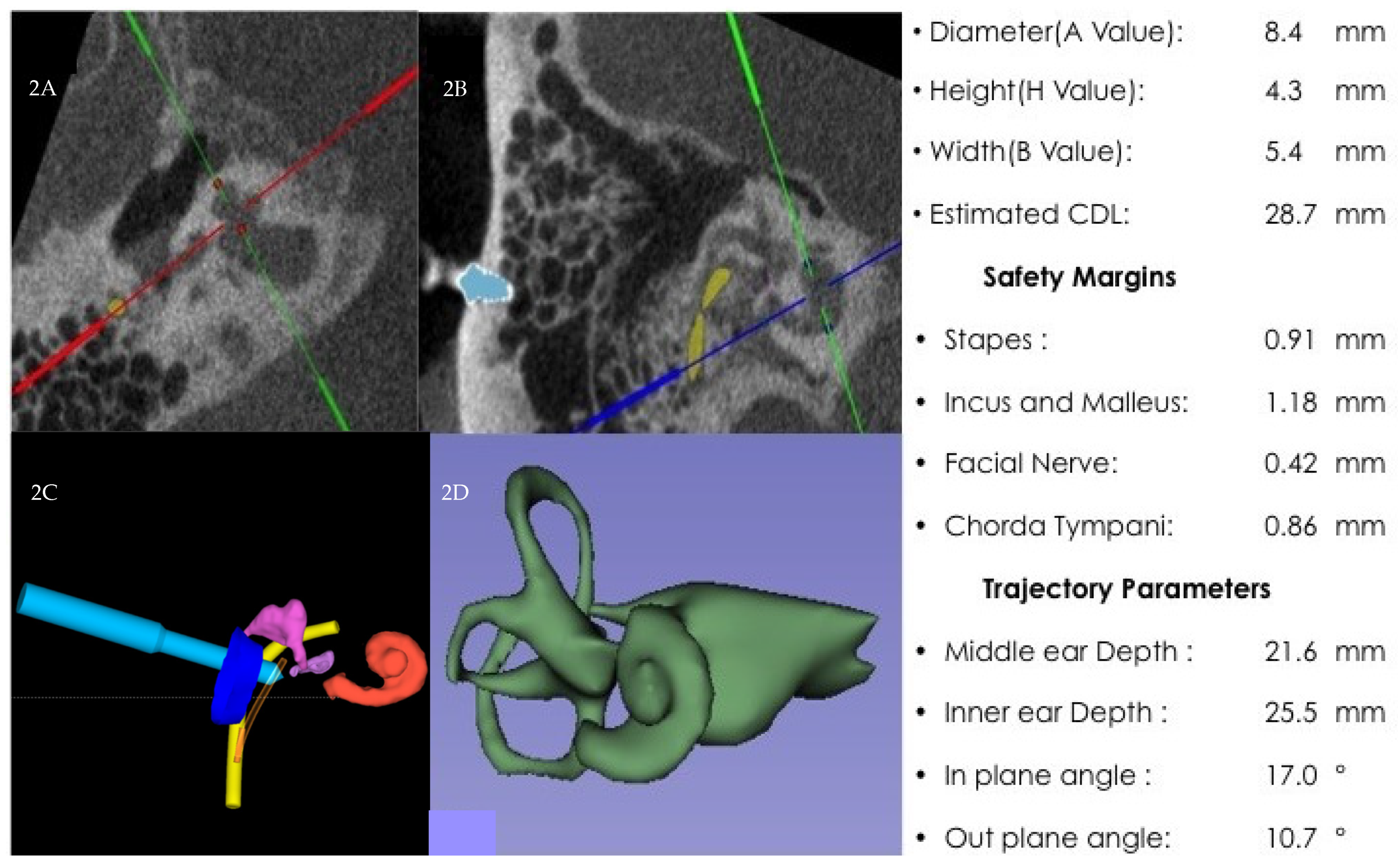



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nucleotide Change | Amino Acide Change | Feature of Deafness | Defects on Temporal Bone CT | Perilymphatic Gusher | Location | Year | References |
|---|---|---|---|---|---|---|---|
| 603–610delCAAA | p.Lys202 fs | SNHL | Yes | The Netherlands | 1995 | [4] | |
| c.648–651delG | p.Arg215 fs | Mixed | Yes | Yes | The Netherlands | 1995 | [4] |
| c.895delA | p.Leu298 fs | Mixed | Yes | Yes | The Netherlands | 1995 | [4] |
| c.950T > G | p.Leu317Trp | Mixed | Yes | The Netherlands | 1995 | [4] | |
| c.1000A > G | p.Lys334Glu | Mixed | Yes | The Netherlands | 1995 | [4] | |
| c.862del4 | p.Ser288Gln | Mixed | Yes | Yes | UK | 1995 | [17] |
| c.935C > T | p.Ala312Val | SNHL | Yes | UK | 1995 | [17] | |
| del 2.6 kb, 6.5 kb | Korea | 1996 | [5] | ||||
| del 7 kb, 4.4 kb | Korea | 1996 | [5] | ||||
| del 8 kb | Mixed | Yes | Korea | 1996 | [5] | ||
| del 20 kb | Mixed | Yes | Korea | 1996 | [5] | ||
| del 130 kb | Mixed | Yes | Korea | 1996 | [5] | ||
| del 200 kb | Mixed | Yes | Korea | 1996 | [5] | ||
| del 220 kb | Mixed | Yes | Korea | 1996 | [5] | ||
| del entire gene | Mixed | Yes | Korea | 1996 | [5] | ||
| c.967C > G | p.Arg323Gly | Mixed | Yes | The Netherlands | 1997 | [10] | |
| c.990A > T | p.Arg330Ser | SNHL | Yes | Yes | The Netherlands | 1997 | [10] |
| c.985C > G | p.Arg329Gly | Mixed | Yes | Yes | US | 1997 | [31] |
| c.689C > T | p.Thr230Ile | Mixed | Yes | Yes | US | 1997 | [31] |
| 601–606delTTCAAA | p.Phe201/Lys202del | Mixed | Yes | Yes | Japan | 1998 | [26] |
| del 1200 kb | SNHL | Yes | Spain | 2000 | [32] | ||
| del530 kb | SNHL | Yes | Yes | US | 2005 | [18] | |
| c. 683C > T | p.Ser228Leu | SNHL | Yes | US | 2005 | [18] | |
| c.925 T > C | p.Ser309Pro | SNHL | Yes | China | 2006 | [19] | |
| c.383delG | p.Gly128fs | SNHL | Yes | Korea | 2009 | [33] | |
| c.623T > A | p.L208Ter | SNHL | Yes | Korea | 2009 | [33] | |
| c.986G > C | p.Arg329Pro | Mixed | Yes | Korea | 2009 | [15] | |
| c.927–929del | p.Ser310del | Mixed | Yes | Korea | 2009 | [15] | |
| c.293C > A | p.Ser98Ter * | Mixed | Yes | France | 2009 | [28] | |
| p.Glu236Asp * | Normal | No | France | 2009 | [28] | ||
| p.Arg282Gln * | Normal | No | France | 2009 | [28] | ||
| p.Ile285Asn * | Normal | France | 2009 | [28] | |||
| p.Ser288CysfsTer40 * | Normal | France | 2009 | [28] | |||
| p.Ile308Asn * | Mixed | No | France | 2009 | [28] | ||
| p.Ile308 IlefsTer28 * | Normal | No | France | 2009 | [28] | ||
| c.499 C > T | p.Arg167Ter | Mixed | Yes | Yes | Korea | 2010 | [34] |
| c.647G > A | p.Gly216 Glu | SNHL | Yes | China | 2010 | [20] | |
| c.973 T > A | p.Trp325Arg | SNHL | Yes | Yes | Germany | 2011 | [35] |
| c.341G > A | p.Trp114Ter | SNHL | Yes | Pakistan | [21] | ||
| c.406C > T | p.Gln136Ter | SNHL | Yes | Pakistan | [21] | ||
| c.235C > T | p.Gln79Ter | SNHL | Yes | Israel | [36] | ||
| c.853 854del | p.Ile285Argfs43Ter | Israel | [36] | ||||
| c.623 T > A | p.Leu208Ter | SNHL | Yes | Korea | 2013 | [8] | |
| c.632C > T | p.Thr211Met | Mixed | Yes | Korea | 2013 | [8] | |
| c.686A > G | p.Gln229Arg | SNHL | Yes | Korea | 2013 | [8] | |
| c.950dupT | p.Leu317PhefsTer12 | SNHL | Yes | Korea | 2013 | [8] | |
| c.1069delA | p.Thr 354GlnfsTer115 | SNHL | Yes | Korea | 2013 | [8] | |
| c.1084 T > C | p.Ter362ArgextTer113 | SNHL | Yes | Korea | 2013 | [8] | |
| c.987 T > C | p.Leu308Thr | Mixed | Nigeria | 2015 | [22] | ||
| c.902C > T | p.Pro301Leu | Mixed | Ecuador | 2015 | [22] | ||
| c.772delG | p. Glu 258Argfs | SNHL | Yes | Turkey | 2015 | [22] | |
| c.707A > C | p.Glu236Ala | SNHL | Turkey | 2015 | [22] | ||
| c.346delG | p.Ala116Profs | SNHL | Turkey | 2015 | [22] | ||
| c.727_728insA | p.Asn244LysfsTer26 | SNHL | Yes | Yes | Japan | 2015 | [27] |
| c.79C > T | p.Gln27Ter | SNHL | Yes | Yes | Poland | 2016 | [37] |
| c.346delG | p.Ala116Profs | SNHL | Yes | Yes | Poland | 2016 | [37] |
| c.559G > T | p.Glu187Ter | SNHL | Yes | Poland | 2016 | [37] | |
| c.623 T > A | p.Leu208Ter | SNHL | Yes | Yes | Poland | 2016 | [37] |
| c.650 T > A | p.Leu217Ter | SNHL | Yes | Yes | Poland | 2016 | [37] |
| c.823C > T | p.Gln275Ter | SNHL | Yes | Yes | Poland | 2016 | [37] |
| c.916C > T | p.Gln306Ter | SNHL | Yes | Poland | 2016 | [37] | |
| c.971 T > A | p.Val324Asp | SNHL | Yes | Yes | Poland | 2016 | [37] |
| c.982A > G | p.Lys328Glu * | SNHL | Taiwan | 2017 | [38] | ||
| c.499C > T | Mixed | Yes | China | 2017 | [39] | ||
| c.927delCTC | p.Ser310del | Mixed | Yes | China | 2018 | [40] | |
| c.669 T > A | p.Tyr223Ter | SNHL | Yes | Yes | China | 2018 | [40] |
| c.973delT | p.Trp325GlyfsTer12 | Mixed | Yes | China | 2018 | [40] | |
| c.975G > A | p.Trp325Ter | Mixed | Yes | Russia | 2018 | [23] | |
| c.852delC | p.Ile285Serfs;Ter3 | Mixed | Yes | Korean | 2019 | [41] | |
| c.76C > T | p.Gln26Ter | Mixed | Yes | China | 2019 | [24] | |
| c.604 A > G | p.Lys202Glu | SNHL | Vietnam | 2019 | [25] | ||
| c.699C > A | p.Cys233Ter | SNHL | Yes | Yes | China | 2020 | [42] |
| c.400_401insACTC | p.Gln136LfsTer58 | SNHL | Yes | Yes | China | 2020 | [29] |
| c.870G > T | p.Lys290Asn | Mixed | Yes | Italy | 2020 | [30] | |
| c.934G > C | p.Ala31Pro * | Mixed | Yes | Yes | Turkey | 2020 | Present Study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tekin, A.M.; Matulic, M.; Wuyts, W.; Assadi, M.Z.; Mertens, G.; Rompaey, V.v.; Li, Y.; Heyning, P.v.d.; Topsakal, V. A New Pathogenic Variant in POU3F4 Causing Deafness Due to an Incomplete Partition of the Cochlea Paved the Way for Innovative Surgery. Genes 2021, 12, 613. https://doi.org/10.3390/genes12050613
Tekin AM, Matulic M, Wuyts W, Assadi MZ, Mertens G, Rompaey Vv, Li Y, Heyning Pvd, Topsakal V. A New Pathogenic Variant in POU3F4 Causing Deafness Due to an Incomplete Partition of the Cochlea Paved the Way for Innovative Surgery. Genes. 2021; 12(5):613. https://doi.org/10.3390/genes12050613
Chicago/Turabian StyleTekin, Ahmet M., Marco Matulic, Wim Wuyts, Masoud Zoka Assadi, Griet Mertens, Vincent van Rompaey, Yongxin Li, Paul van de Heyning, and Vedat Topsakal. 2021. "A New Pathogenic Variant in POU3F4 Causing Deafness Due to an Incomplete Partition of the Cochlea Paved the Way for Innovative Surgery" Genes 12, no. 5: 613. https://doi.org/10.3390/genes12050613