Non-Penetrance for Ocular Phenotype in Two Individuals Carrying Heterozygous Loss-of-Function ZEB1 Alleles

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
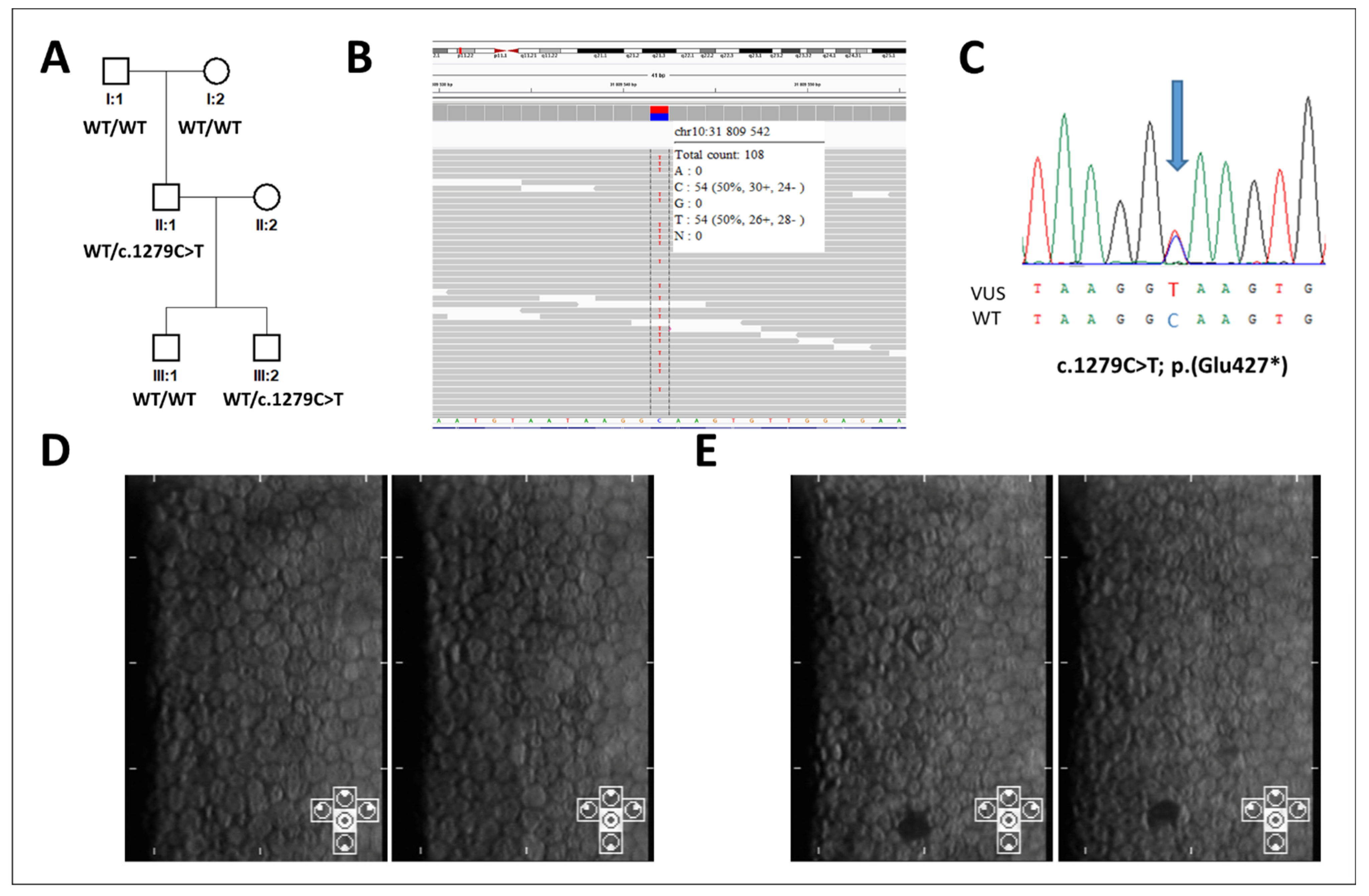
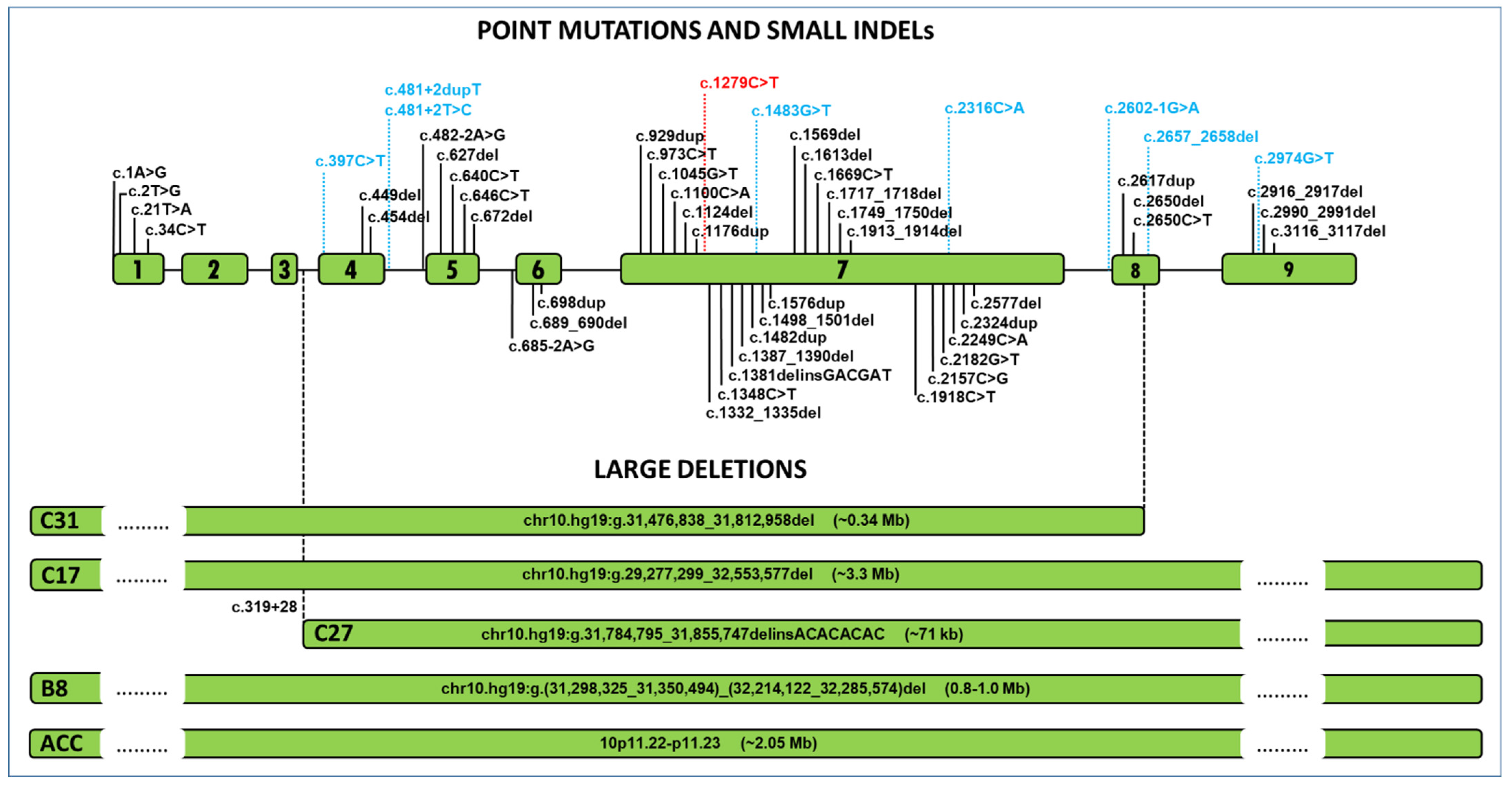
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Krachmer, J.H. Posterior polymorphous corneal dystrophy: A disease characterized by epithelial-like endothelial cells which influence management and prognosis. Trans. Am. Ophthalmol. Soc. 1985, 83, 413–475. [Google Scholar]
- Liskova, P.; Palos, M.; Hardcastle, A.J.; Vincent, A.L. Further genetic and clinical insights of posterior polymorphous corneal dystrophy 3. JAMA Ophthalmol. 2013, 131, 1296–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liskova, P.; Dudakova, L.; Evans, C.J.; Rojas Lopez, K.E.; Pontikos, N.; Athanasiou, D.; Jama, H.; Sach, J.; Skalicka, P.; Stranecky, V.; et al. Ectopic GRHL2 Expression Due to Non-coding Mutations Promotes Cell State Transition and Causes Posterior Polymorphous Corneal Dystrophy 4. Am. J. Hum. Genet. 2018, 102, 447–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef] [PubMed]
- Plygawko, A.T.; Kan, S.; Campbell, K. Epithelial-mesenchymal plasticity: Emerging parallels between tissue morphogenesis and cancer metastasis. Philos. Trans. R Soc. Lond. B Biol. Sci. 2020, 375, 20200087. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.E.; Liskova, P.; Evans, C.J.; Dudakova, L.; Noskova, L.; Pontikos, N.; Hartmanova, H.; Hodanova, K.; Stranecky, V.; Kozmik, Z.; et al. Autosomal-Dominant Corneal Endothelial Dystrophies CHED1 and PPCD1 Are Allelic Disorders Caused by Non-coding Mutations in the Promoter of OVOL2. Am. J. Hum. Genet. 2016, 98, 75–89. [Google Scholar] [CrossRef] [Green Version]
- Krafchak, C.M.; Pawar, H.; Moroi, S.E.; Sugar, A.; Lichter, P.R.; Mackey, D.A.; Mian, S.; Nairus, T.; Elner, V.; Schteingart, M.T.; et al. Mutations in TCF8 cause posterior polymorphous corneal dystrophy and ectopic expression of COL4A3 by corneal endothelial cells. Am. J. Hum. Genet. 2005, 77, 694–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dudakova, L.; Evans, C.J.; Pontikos, N.; Hafford-Tear, N.J.; Malinka, F.; Skalicka, P.; Horinek, A.; Munier, F.L.; Voide, N.; Studeny, P.; et al. The utility of massively parallel sequencing for posterior polymorphous corneal dystrophy type 3 molecular diagnosis. Exp. Eye Res. 2019, 182, 160–166. [Google Scholar] [CrossRef]
- Cunnusamy, K.; Bowman, C.B.; Beebe, W.; Gong, X.; Hogan, R.N.; Mootha, V.V. Congenital Corneal Endothelial Dystrophies Resulting from Novel De Novo Mutations. Cornea 2016, 35, 281–285. [Google Scholar] [CrossRef] [Green Version]
- Liskova, P.; Evans, C.J.; Davidson, A.E.; Zaliova, M.; Dudakova, L.; Trkova, M.; Stranecky, V.; Carnt, N.; Plagnol, V.; Vincent, A.V.; et al. Heterozygous deletions at the ZEB1 locus verify haploinsufficiency as the mechanism of disease for posterior polymorphous corneal dystrophy type 3. Eur. J. Hum. Genet. 2016, 24, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Liskova, P.; Filipec, M.; Merjava, S.; Jirsova, K.; Tuft, S.J. Variable ocular phenotypes of posterior polymorphous corneal dystrophy caused by mutations in the ZEB1 gene. Ophthalmic Genet. 2010, 31, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.S.; Roldan, A.N.; Frausto, R.F.; Aldave, A.J. Posterior polymorphous corneal dystrophy 3 is associated with agenesis and hypoplasia of the corpus callosum. Vis. Res. 2014, 100, 88–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaudhry, A.; Chung, B.H.; Stavropoulos, D.J.; Araya, M.P.; Ali, A.; Heon, E.; Chitayat, D. Agenesis of the corpus callosum, developmental delay, autism spectrum disorder, facial dysmorphism, and posterior polymorphous corneal dystrophy associated with ZEB1 gene deletion. Am. J. Med. Genet. A. 2017, 173, 2467–2471. [Google Scholar] [CrossRef]
- Collins, R.L.; Brand, H.; Karczewski, K.J.; Zhao, X.; Alfoldi, J.; Francioli, L.C.; Khera, A.V.; Lowther, C.; Gauthier, L.D.; Wang, H.; et al. A structural variation reference for medical and population genetics. Nature 2020, 581, 444–451. [Google Scholar] [CrossRef]
- Galgauskas, S.; Norvydaite, D.; Krasauskaite, D.; Stech, S.; Asoklis, R.S. Age-related changes in corneal thickness and endothelial characteristics. Clin. Interv. Aging 2013, 8, 1445–1450. [Google Scholar] [CrossRef] [Green Version]
- Zoega, G.M.; Fujisawa, A.; Sasaki, H.; Kubota, A.; Sasaki, K.; Kitagawa, K.; Jonasson, F. Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study. Ophthalmology 2006, 113, 565–569. [Google Scholar] [CrossRef]
- Higa, A.; Sakai, H.; Sawaguchi, S.; Iwase, A.; Tomidokoro, A.; Amano, S.; Araie, M. Prevalence of and risk factors for cornea guttata in a population-based study in a southwestern island of Japan: The Kumejima study. Arch. Ophthalmol. 2011, 129, 332–336. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Evans, C.J.; Liskova, P.; Dudakova, L.; Hrabcikova, P.; Horinek, A.; Jirsova, K.; Filipec, M.; Hardcastle, A.J.; Davidson, A.E.; Tuft, S.J. Identification of six novel mutations in ZEB1 and description of the associated phenotypes in patients with posterior polymorphous corneal dystrophy 3. Ann. Hum. Genet. 2015, 79, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Wang, J.; Tian, X.; Yu, H.; Truong, C.; Mitchell, J.J.; Wierenga, K.J.; Craigen, W.J.; Zhang, V.W.; Wong, L.C. Detection and Quantification of Mosaic Mutations in Disease Genes by Next-Generation Sequencing. J. Mol. Diagn. 2016, 18, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Palmer, E.E.; Mowat, D. Agenesis of the corpus callosum: A clinical approach to diagnosis. Am. J. Med. Genet. C Semin. Med. Genet. 2014, 166C, 184–197. [Google Scholar] [CrossRef] [PubMed]
- Liskova, P.; Tuft, S.J.; Gwilliam, R.; Ebenezer, N.D.; Jirsova, K.; Prescott, Q.; Martincova, R.; Pretorius, M.; Sinclair, N.; Boase, D.L.; et al. Novel mutations in the ZEB1 gene identified in Czech and British patients with posterior polymorphous corneal dystrophy. Hum. Mutat. 2007, 28, 638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, A.; Colin, E.; Goudenège, D.; Bonneau, D. A spanshot of some pLI score pitfalls. Hum. Mutat. 2019, 40, 839–841. [Google Scholar]



{kind=link}
{kind=link}
| Individual ID | Age (Years)/Gender | BCVA | Refractive ErrorDS/DC | ECD (Cells/mm2) | |||
|---|---|---|---|---|---|---|---|
| RE | LE | RE | LE | RE | LE | ||
| II:1 | 47/M | 1.0 | 1.0 | −3.25/−0.5 × 180° | −3.0/−1.25 × 135° | 2538 | 2638 |
| III:2 | 7/M | 1.0 | 1.0 | +0.25/−0.75 × 179° | −/−0.25 × 171° | 3355 | 3125 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dudakova, L.; Stranecky, V.; Piherova, L.; Palecek, T.; Pontikos, N.; Kmoch, S.; Skalicka, P.; Vaneckova, M.; Davidson, A.E.; Liskova, P. Non-Penetrance for Ocular Phenotype in Two Individuals Carrying Heterozygous Loss-of-Function ZEB1 Alleles. Genes 2021, 12, 677. https://doi.org/10.3390/genes12050677
Dudakova L, Stranecky V, Piherova L, Palecek T, Pontikos N, Kmoch S, Skalicka P, Vaneckova M, Davidson AE, Liskova P. Non-Penetrance for Ocular Phenotype in Two Individuals Carrying Heterozygous Loss-of-Function ZEB1 Alleles. Genes. 2021; 12(5):677. https://doi.org/10.3390/genes12050677
Chicago/Turabian StyleDudakova, Lubica, Viktor Stranecky, Lenka Piherova, Tomas Palecek, Nikolas Pontikos, Stanislav Kmoch, Pavlina Skalicka, Manuela Vaneckova, Alice E. Davidson, and Petra Liskova. 2021. "Non-Penetrance for Ocular Phenotype in Two Individuals Carrying Heterozygous Loss-of-Function ZEB1 Alleles" Genes 12, no. 5: 677. https://doi.org/10.3390/genes12050677