
L2HGDH Missense Variant in a Cat with L-2-Hydroxyglutaric Aciduria

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Clinical Examination
2.3. Magnetic Resonance Imaging
2.4. Metabolic Screening
2.5. Control Samples for Genetic Analyses
2.6. DNA Extraction
2.7. Whole-Genome Sequencing
2.8. Variant Calling
2.9. Gene Analysis
2.10. In Silico Functional Predictions and Database Searches
2.11. PCR and Sanger Sequencing
3. Results
3.1. Clinical History and Examination
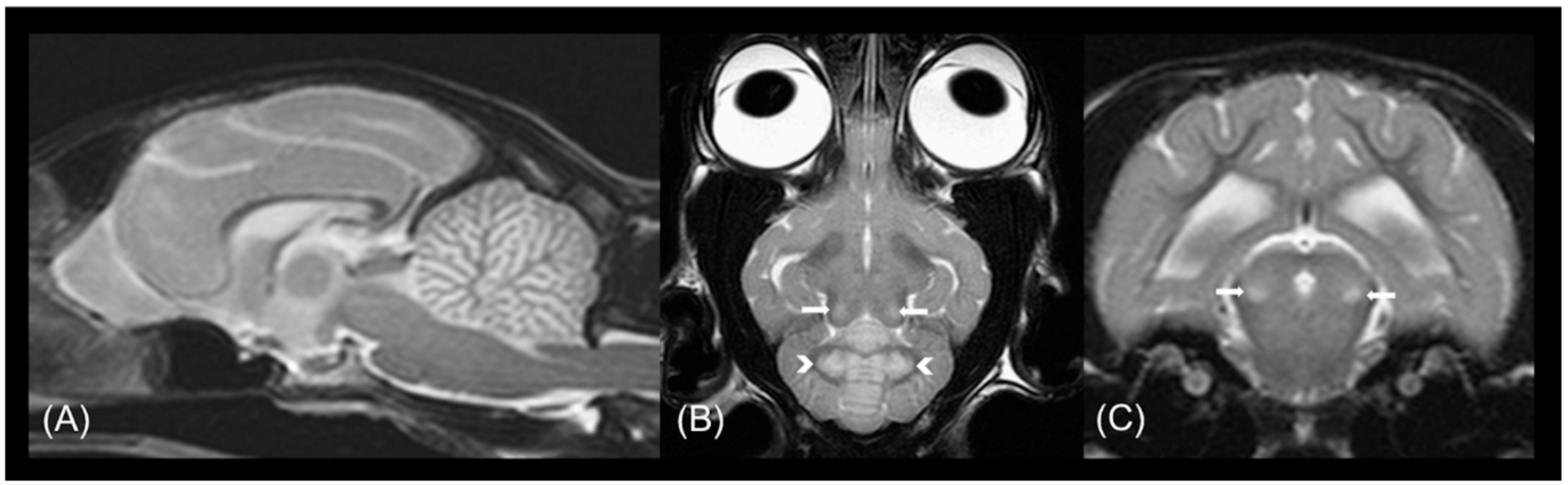
3.2. Radiological Examination
3.3. Laboratory Findings
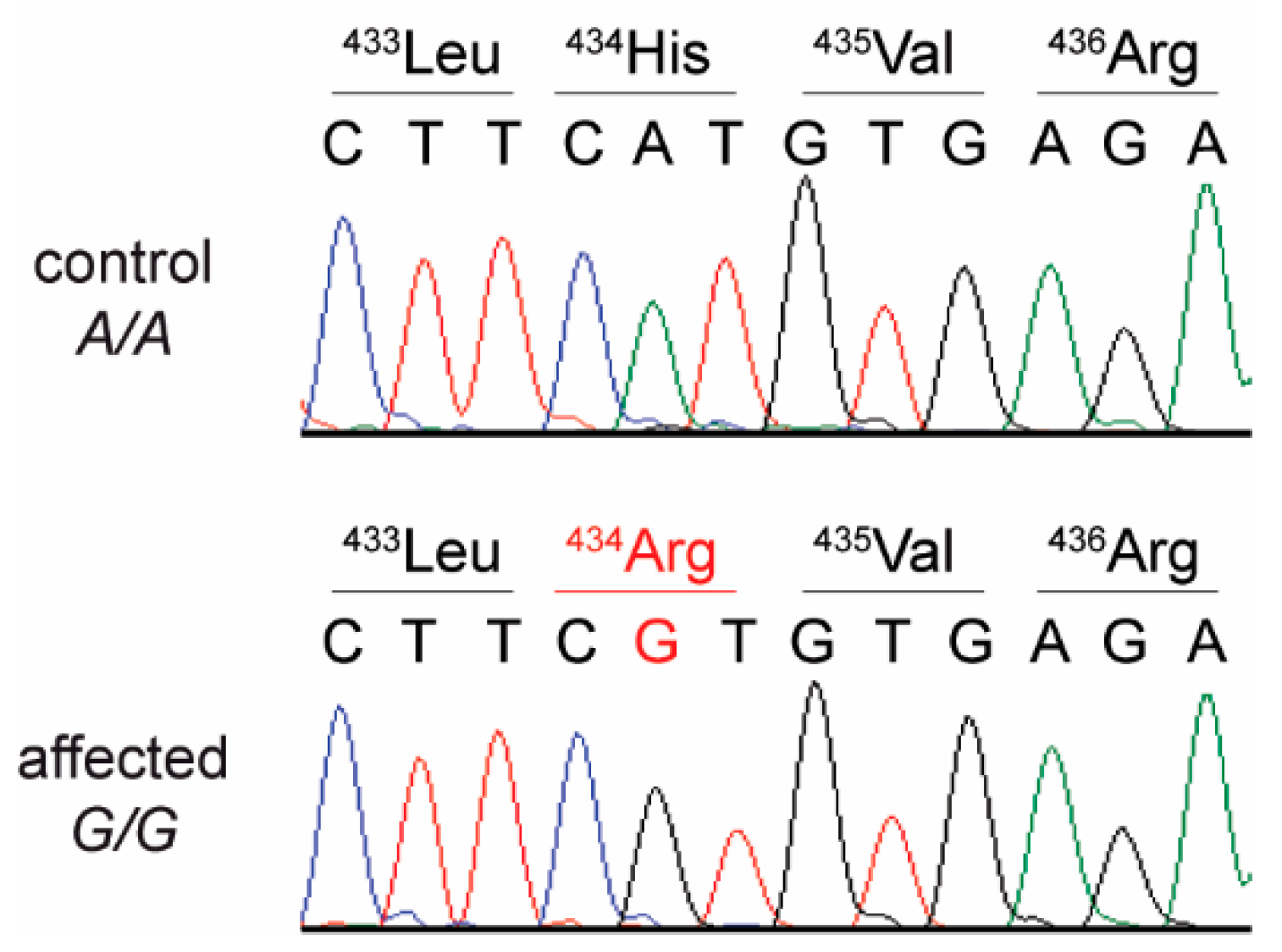
3.4. Genetic Analysis
3.5. Treatment and Outcome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Duran, M.; Kamerling, J.P.; Bakker, H.D.; van Gennip, A.H.; Wadman, S.K. L-2-Hydroxyglutaric aciduria: An inborn error of metabolism? J. Inherit. Metab. Dis. 1980, 3, 109–112. [Google Scholar] [CrossRef]
- Abramson, C.J.; Platt, S.R.; Jakobs, C.; Verhoeven, N.M.; Dennis, R.; Garosi, L.; Diane Shelton, G. L-2-Hydroxyglutaric Aciduria in Staffordshire Bull Terriers. J. Vet. Intern. Med. 2003, 17, 551–556. [Google Scholar] [CrossRef]
- Garosi, L.S.; Penderis, J.; McConnell, J.F.; Jakobs, C. L-2-hydroxyglutaric aciduria in a West Highland white terrier. Vet. Rec. 2005, 156, 145–147. [Google Scholar] [CrossRef]
- Sanchez-Masian, D.F.; Artuch, R.; Mascort, J.; Jakobs, C.; Salomons, G.; Zamora, A.; Casado, M.; Fernandez, M.; Recio, A.; Lujan, A. L-2-hydroxyglutaric aciduria in two female Yorkshire terriers. J. Am. Anim. Hosp. Assoc. 2012, 48, 366–371. [Google Scholar] [CrossRef]
- Nye, G.J.; Major, A.C.; Liebel, F.X. 2-Hydroxyglutaric aciduria as a cause for seizure-like episodes in a domestic shorthair cat. J. Feline Med. Surg. Open Rep. 2019, 5, 2055116919853898. [Google Scholar] [CrossRef]
- Shea, A.; De Risio, L.; Carruthers, H.; Ekiri, A.; Beltran, E. Clinical features and disease progression of L-2-hydroxyglutaric aciduria in 27 Staffordshire bull terriers. Vet. Rec. 2016, 179. [Google Scholar] [CrossRef] [PubMed]
- Steenweg, M.E.; Salomons, G.S.; Yapici, Z.; Uziel, G.; Scalais, E.; Zafeiriou, D.I.; Ruiz-Falco, M.L.; Mejaški-Bošnjak, V.; Augoustides-Savvopoulou, P.; Wajner, M.; et al. L-2-Hydroxyglutaric Aciduria: Pattern of MR Imaging Abnormalities in 56 Patients. Radiology 2009, 251, 856–865. [Google Scholar] [CrossRef]
- Topçu, M.; Jobard, F.; Halliez, S.; Coskun, T.; Yalçinkayal, C.; Gerceker, F.O.; Wanders, R.J.A.; Prud’homme, J.F.; Lathrop, M.; Özguc, M.; et al. L-2-hydroxyglutaric aciduria: Identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum. Mol. Genet. 2004, 13, 2803–2811. [Google Scholar] [CrossRef]
- Steenweg, M.E.; Jakobs, C.; Errami, A.; van Dooren, S.J.M.; Adeva Bartolomé, M.T.; Aerssens, P.; Augoustides-Savvapoulou, P.; Baric, I.; Baumann, M.; Bonafé, L.; et al. An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: A genotype-phenotype study. Hum. Mutat. 2010, 31, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Penderis, J.; Calvin, J.; Abramson, C.; Jakobs, C.; Pettitt, L.; Binns, M.M.; Verhoeven, N.M.; O’Driscoll, E.; Platt, S.R.; Mellersh, C.S. L-2-hydroxyglutaric aciduria: Characterisation of the molecular defect in a spontaneous canine model. J. Med. Genet. 2007, 44, 334–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farias, F.H.G.; Zeng, R.; Johnson, G.S.; Shelton, G.D.; Paquette, D.; O’Brien, D.P. A L2HGDH initiator methionine codon mutation in a Yorkshire terrier with L-2-hydroxyglutaric aciduria. BMC Vet. Res. 2012, 8, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rzem, R.; Achouri, Y.; Marbaix, E.; Schakman, O.; Wiame, E.; Marie, S.; Gailly, P.; Vincent, M.F.; Veiga-Da-cunha, M.; Van Schaftingen, E. A mouse model of L-2-hydroxyglutaric aciduria, a disorder of metabolite repair. PLoS ONE 2015, 10. [Google Scholar] [CrossRef]
- Ma, S.; Sun, R.; Jiang, B.; Gao, J.; Deng, W.; Liu, P.; He, R.; Cui, J.; Ji, M.; Yi, W.; et al. L2hgdh Deficiency Accumulates l-2-Hydroxyglutarate with Progressive Leukoencephalopathy and Neurodegeneration. Mol. Cell. Biol. 2017, 37, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaftingen, E.; Rzem, R.; Veiga-da-Cunha, M. L-2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J. Inherit. Metab. Dis. 2009, 32, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Intlekofer, A.M.; DeMatteo, R.G.; Venneti, S.; Finley, L.W.S.; Lu, C.; Judkins, A.R.; Rustenburg, A.S.; Grinaway, P.B.; Chodera, J.D.; Cross, J.R.; et al. Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oldham, W.M.; Clish, C.B.; Yang, Y.; Loscalzo, J. Hypoxi-mediated increasesin L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 2015, 22, 291–303. [Google Scholar] [CrossRef] [Green Version]
- Rzem, R.; Veiga-Da-Cunha, M.; Noël, G.; Goffette, S.; Nassogne, M.C.; Tabarki, B.; Schöller, C.; Marquardt, T.; Vikkula, M.; Van Schaftingen, E. A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc. Natl. Acad. Sci. USA 2004, 101, 16849–16854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brauburger, K.; Burckhardt, G.; Burckhardt, B.C. The sodium-dependent di- and tricarboxylate transporter, NaCT, is not responsible for the uptake of D-, L-2-hydroxyglutarate and 3-hydroxyglutarate into neurons. J. Inherit. Metab. Dis. 2011, 34, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Yang, H.; Liu, Y.; Yang, Y.; Wang, P.; Kim, S.-H.; Ito, S.; Yang, C.; Wang, P.; Xiao, M.-T.; et al. Oncometabolite 2-Hydroxyglutarate Is a Competitive Inhibitor of α-Ketoglutarate-Dependent Dioxygenases. Cancer Cell 2011, 19, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.M.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Veiga-da-Cunha, M.; Van Schaftingen, E.; Bommer, G.T. Inborn errors of metabolite repair. J. Inherit. Metab. Dis. 2020, 43, 14–24. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, R.T.; Zanatta, Â.; Amaral, A.U.; Leipnitz, G.; de Oliveira, F.H.; Seminotti, B.; Wajner, M. Experimental evidence that in vivo intracerebral administration of L-2-hydroxyglutaric acid to neonatal rats provokes disruption of redox status and histopathological abnormalities in the brain. Neurotox. Res. 2018, 33, 681–692. [Google Scholar] [CrossRef]
- Hunt, R.J.; Granat, L.; McElroy, G.S.; Ranganathan, R.; Chandel, N.S.; Bateman, J.M. Mitochondrial stress causes neuronal dysfunction via an ATF4-dependent increase in L-2-hydroxyglutarate. J. Cell Biol. 2019, 218, 4007–4016. [Google Scholar] [CrossRef] [Green Version]
- Sweetman, L. Organic acid analysis. In Techniques in Diagnostic Human Biochemical Genetics: A Laboratory Manual; Hommes, F.A., Ed.; Wiley-Liss: New York, NY, USA, 1991; pp. 143–176. ISBN 047156818X. [Google Scholar]
- Struys, E.A.; Jansen, E.E.W.; Verhoeven, N.M.; Jakobs, C. Measurement of urinary D- and L-2-hydroxyglutarate enantiomers by stable-isotope-dilution liquid chromatography-tandem mass spectrometry after derivatization with diacetyl-L-tartaric anhydride. Clin. Chem. 2004, 50, 1391–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, V.; Drögemüller, C.; Leeb, T.; Aguirre, G.; André, C.; Bannasch, D.; Becker, D.; Davis, B.; Ekenstedt, K.; Faller, K.; et al. A comprehensive biomedical variant catalogue based on whole genome sequences of 582 dogs and eight wolves. Anim. Genet. 2019, 50, 695–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly Austin 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bendl, J.; Stourac, J.; Salanda, O.; Pavelka, A.; Wieben, E.D.; Zendulka, J.; Brezovsky, J.; Damborsky, J. PredictSNP: Robust and Accurate Consensus Classifier for Prediction of Disease-Related Mutations. PLoS Comput. Biol. 2014, 10, 1–11. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef] [Green Version]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. Inferring the molecular and phenotypic impact of amino acid variants with MutPred2. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Online Mendelian Inheritance in Animals, OMIA. Sydney School of Veterinary Science. Available online: https://omia.org/ (accessed on 21 January 2021).
- Côté, E. Clinical Veterinary Advisor: Dogs and Cats, 3rd ed.; Elsevier: St. Louis, MO, USA, 2015; p. 178. [Google Scholar]

{kind=link}
{kind=link}
| Filtering Step | Homozygous Variants | Heterozygous Variants |
|---|---|---|
| All variants in the affected cat | 4,038,732 | 4,552,718 |
| Private variants | 11,860 | 69,034 |
| Protein-changing private variants | 61 | 336 |
| Private variants in L2HGDH candidate gene | 1 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Christen, M.; Janzen, N.; Fraser, A.; Sewell, A.C.; Jagannathan, V.; Guevar, J.; Leeb, T.; Sanchez-Masian, D. L2HGDH Missense Variant in a Cat with L-2-Hydroxyglutaric Aciduria. Genes 2021, 12, 682. https://doi.org/10.3390/genes12050682
Christen M, Janzen N, Fraser A, Sewell AC, Jagannathan V, Guevar J, Leeb T, Sanchez-Masian D. L2HGDH Missense Variant in a Cat with L-2-Hydroxyglutaric Aciduria. Genes. 2021; 12(5):682. https://doi.org/10.3390/genes12050682
Chicago/Turabian StyleChristen, Matthias, Nils Janzen, Anne Fraser, Adrian C. Sewell, Vidhya Jagannathan, Julien Guevar, Tosso Leeb, and Daniel Sanchez-Masian. 2021. "L2HGDH Missense Variant in a Cat with L-2-Hydroxyglutaric Aciduria" Genes 12, no. 5: 682. https://doi.org/10.3390/genes12050682
APA StyleChristen, M., Janzen, N., Fraser, A., Sewell, A. C., Jagannathan, V., Guevar, J., Leeb, T., & Sanchez-Masian, D. (2021). L2HGDH Missense Variant in a Cat with L-2-Hydroxyglutaric Aciduria. Genes, 12(5), 682. https://doi.org/10.3390/genes12050682