
Genome-Wide Association Study Identified Novel Candidate Loci/Genes Affecting Lodging Resistance in Rice

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Field Trials and Trait Measurements
2.3. Genotyping
2.4. Population Structure and Kinship Analysis
2.5. Linkage Disequilibrium Analysis
2.6. Genome-Wide Association Study and Candidate Genes Identification
3. Results
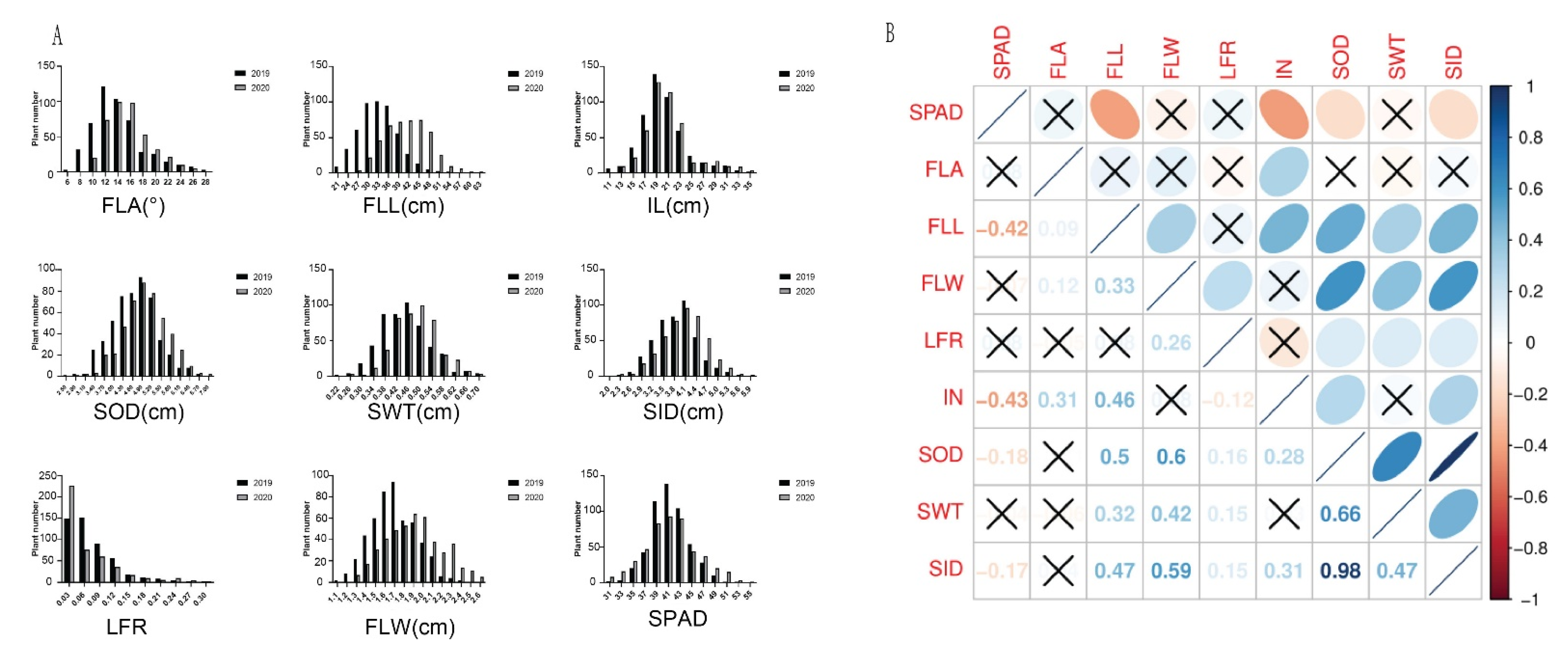
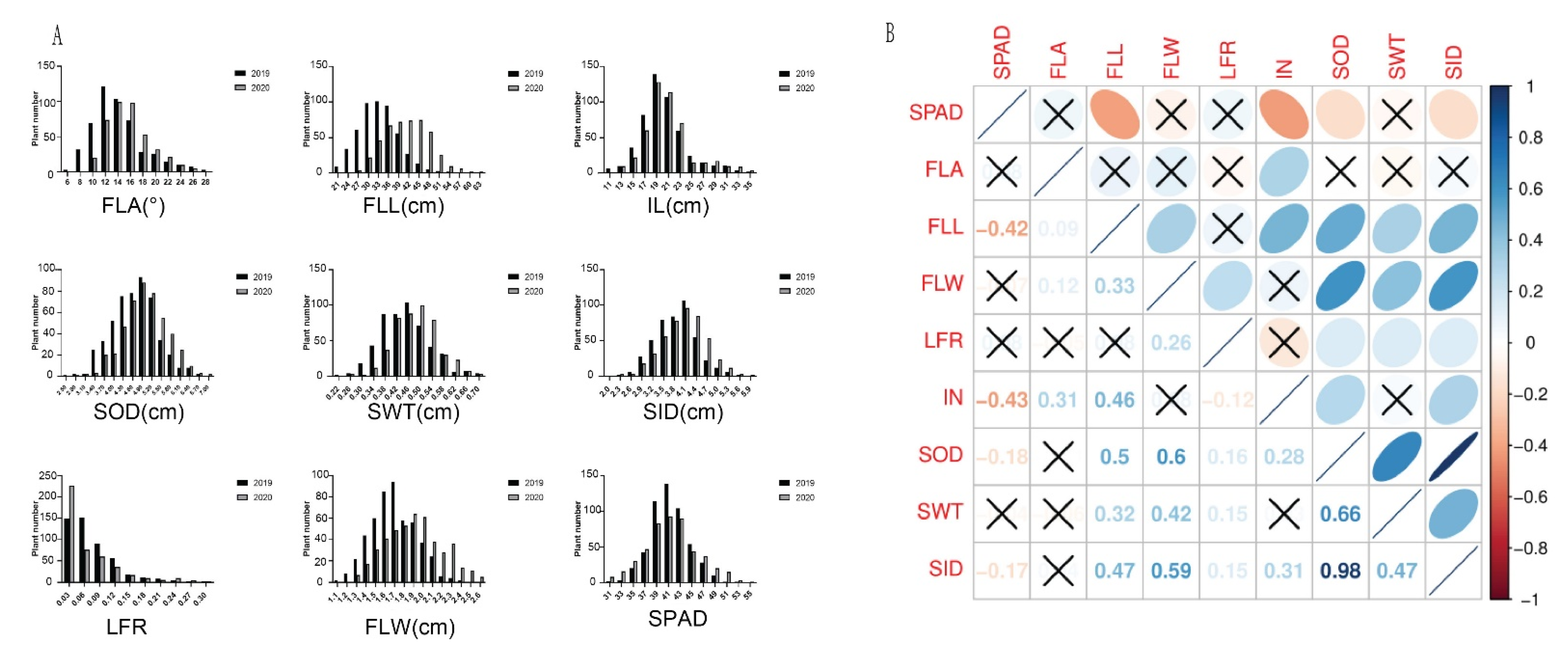
3.1. Phenotypic Variation and Correlations
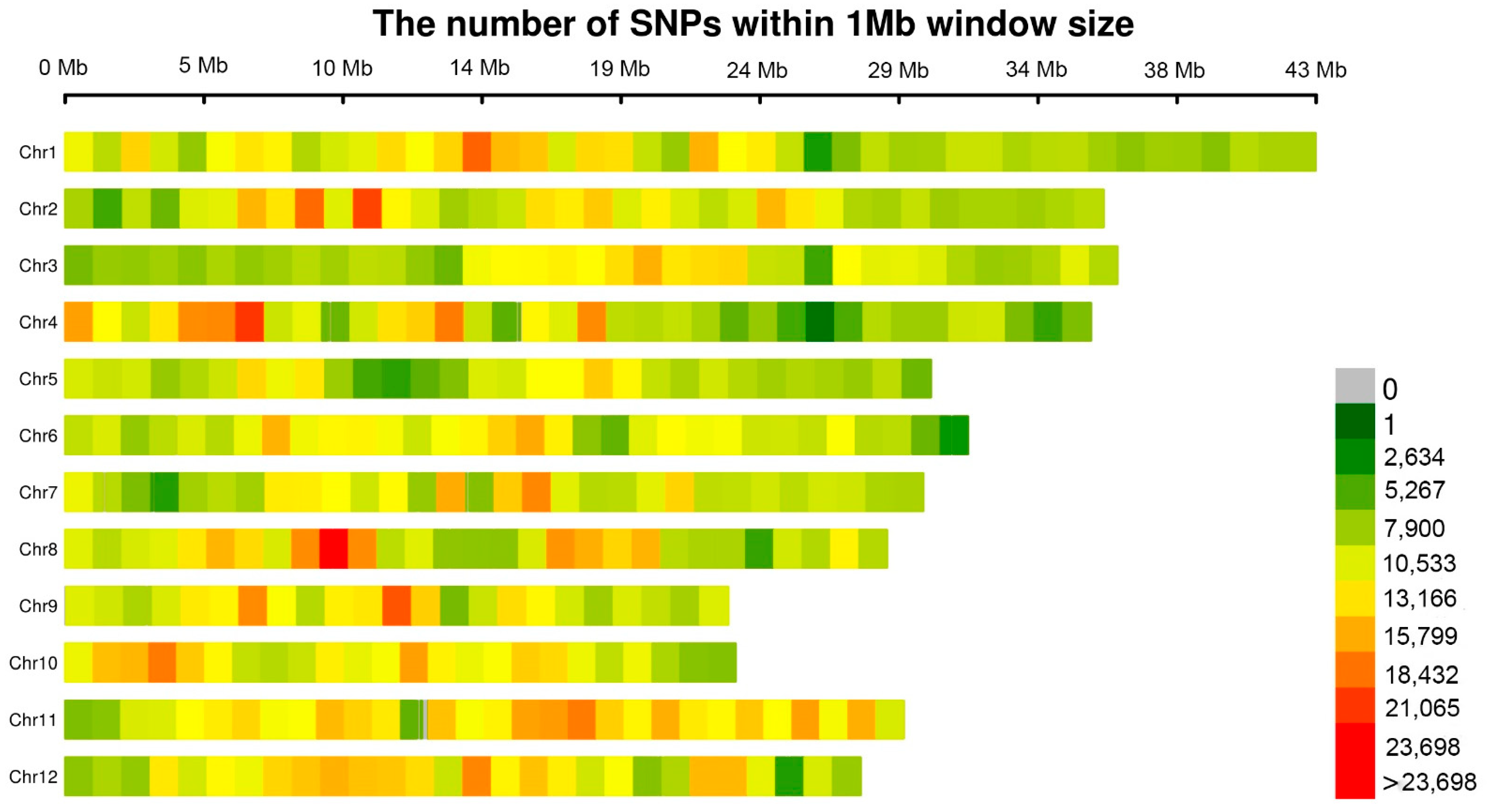
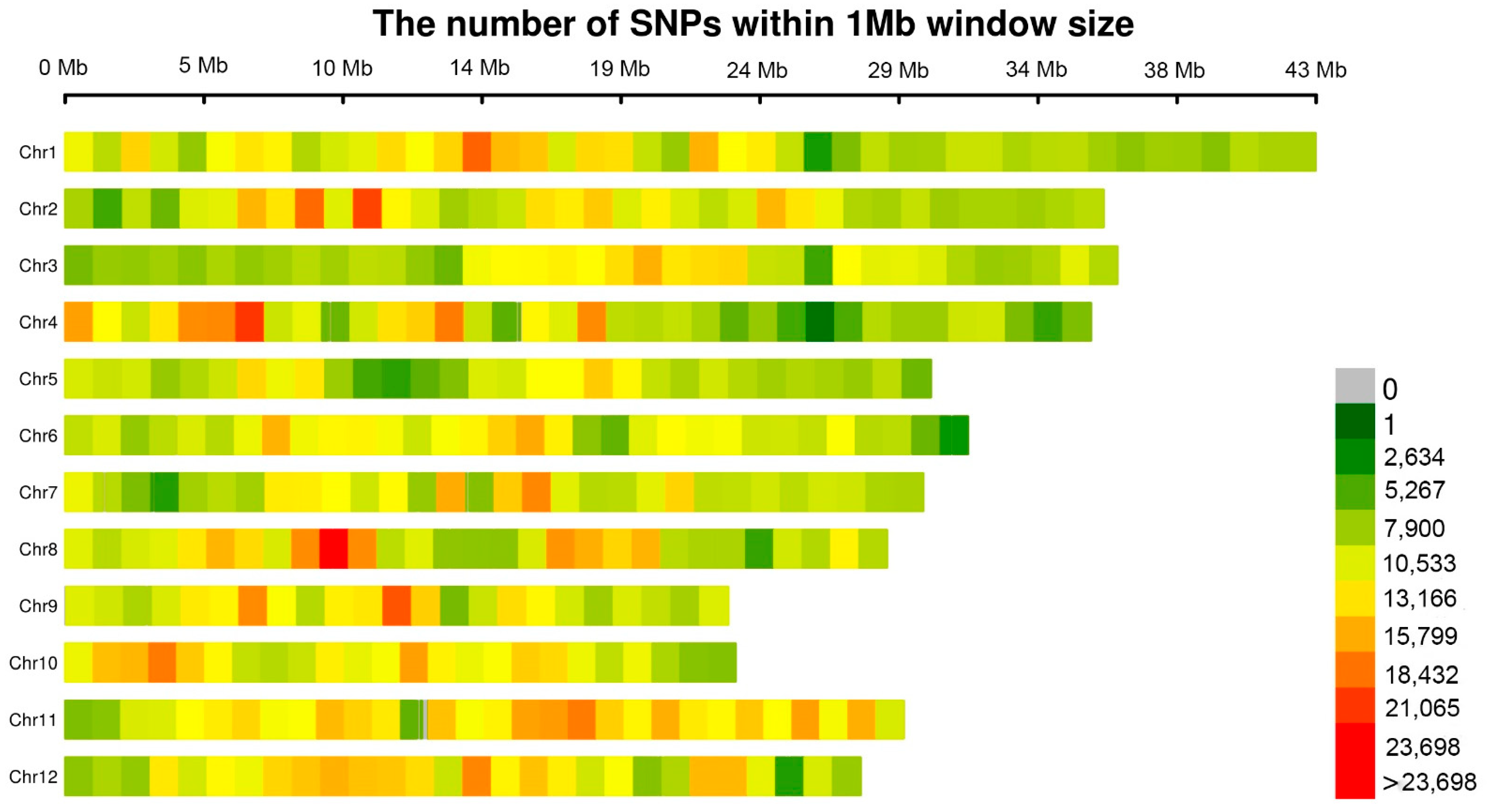
3.2. Statistics of Markers
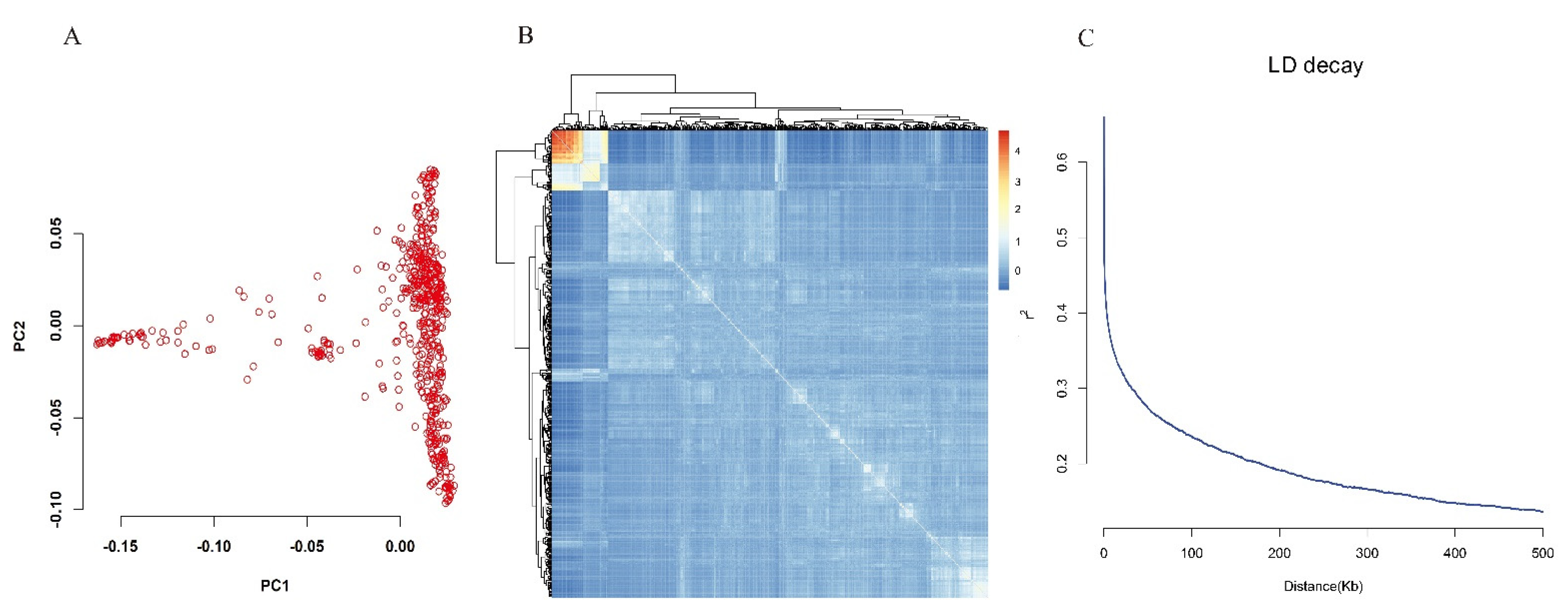
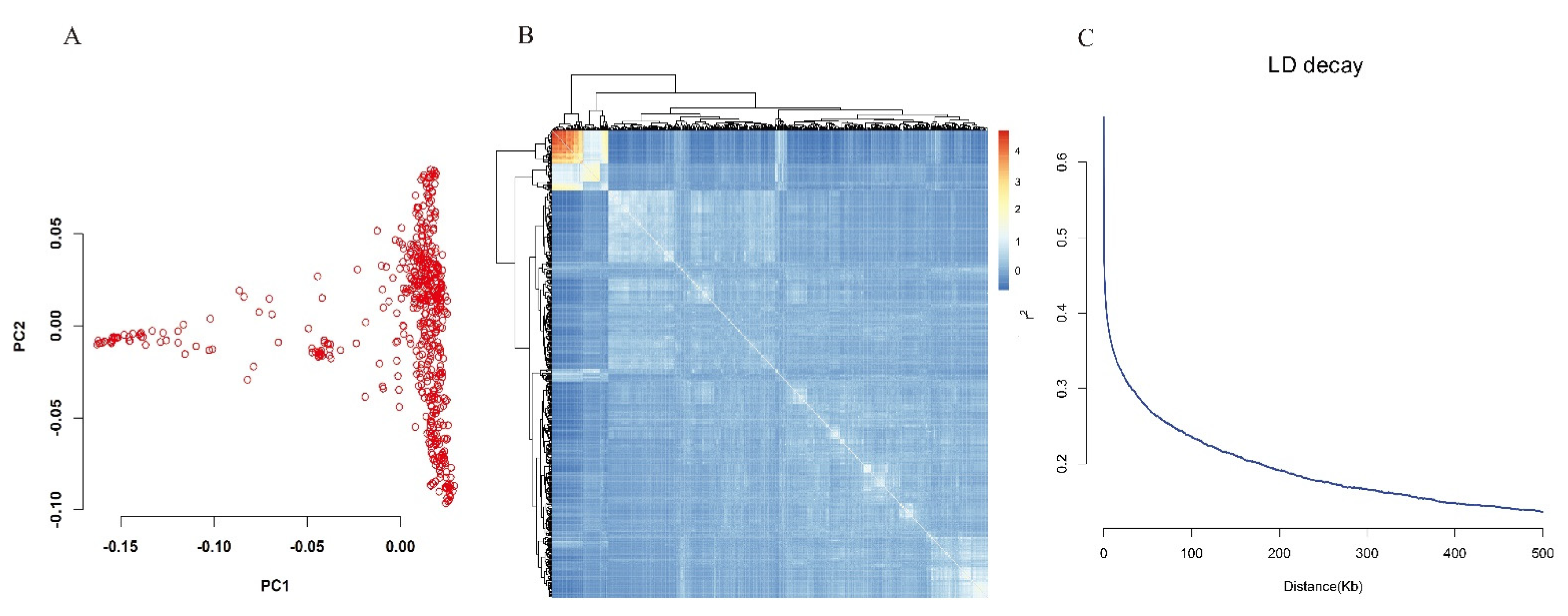
3.3. Population Structure, Kinship and LD Patterns
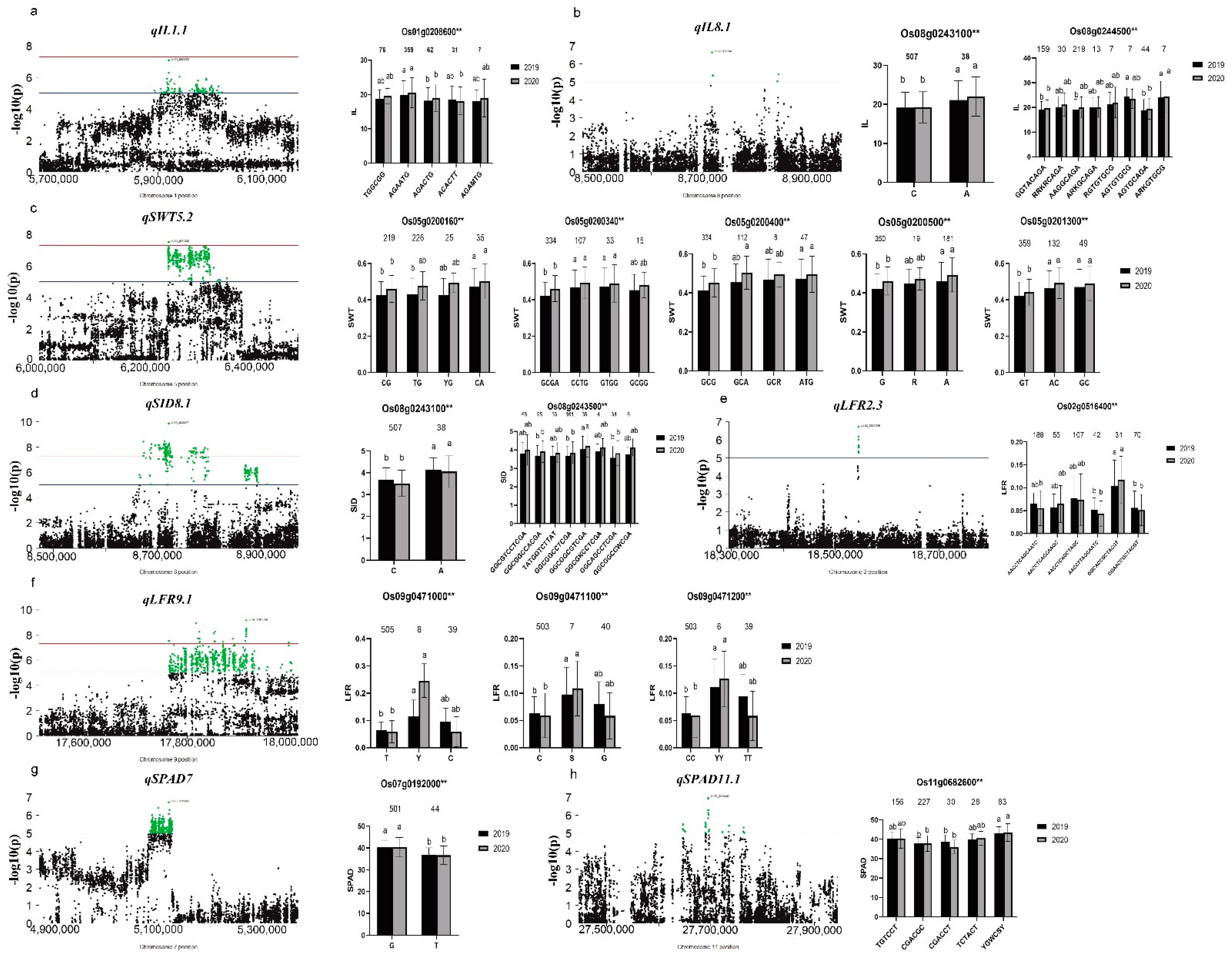
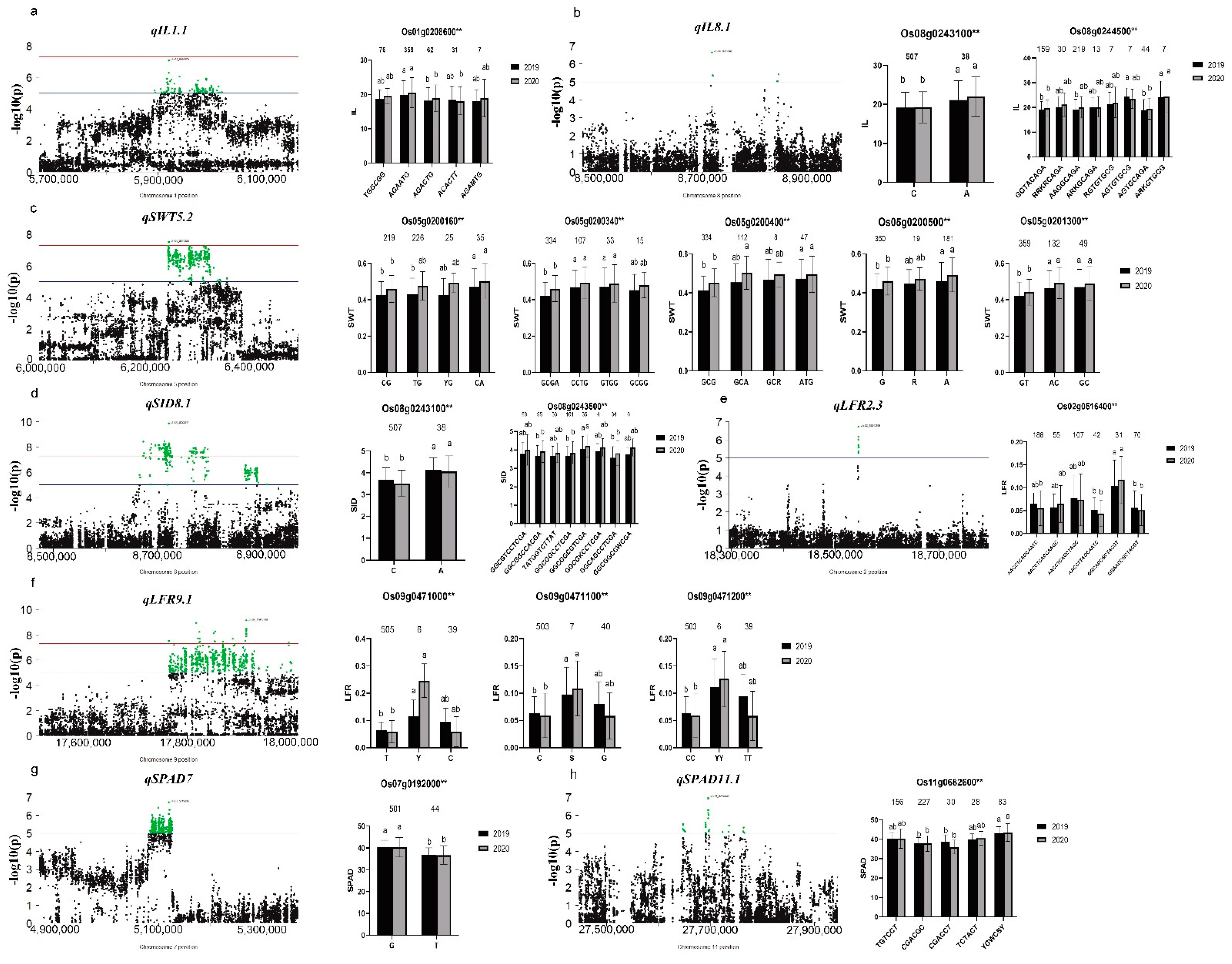
3.4. Identification of Significant Loci for Related Traits through GWAS
3.5. Screening and Haplotype Analysis of Candidate Genes in Important QTL Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berry, P.; Sterling, M.; Spink, J.; Baker, C.; Sylvester-Bradley, R.; Mooney, S.; Tams, A.; Ennos, A. Understanding and Reducing Lodging in Cereals. Adv. Agron. 2004, 84, 217–271. [Google Scholar] [CrossRef]
- Islam, M.S.; Peng, S.; Visperas, R.M.; Ereful, N.; Bhuiya, M.S.U.; Julfiquar, A. Lodging-related morphological traits of hybrid rice in a tropical irrigated ecosystem. Field Crop. Res. 2007, 101, 240–248. [Google Scholar] [CrossRef]
- Peng, J.; Richards, D.E.; Hartley, N.M.; Murphy, G.P.; Devos, K.M.; Flintham, J.E.; Beales, J.; Fish, L.J.; Worland, A.J.; Pelica, F.; et al. ‘Green revolution’ genes encode mutant gibberellin response modulators. Nature 1999, 400, 256–261. [Google Scholar] [CrossRef]
- Sasaki, A.; Ashikari, M.; Ueguchi-Tanaka, M.; Itoh, H.; Nishimura, A.; Swapan, D.; Ishiyama, K.; Saito, T.; Kobayashi, M.; Khush, G.S.; et al. A mutant gibberellin-synthesis gene in rice. Nature 2002, 416, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Takamure, I.; Sato, S.; Tokutome, T.; Sato, Y. Effects of the Dwarfing Gene Originating from ‘Dee-geo-woo-gen’ on Yield and its Related Traits in Rice. Breed. Sci. 2002, 52, 95–100. [Google Scholar] [CrossRef] [Green Version]
- Hirano, K.; Ordonio, R.L.; Matsuoka, M. Engineering the lodging resistance mechanism of post-Green Revolution rice to meet future demands. Proc. Jpn. Acad. Ser. B 2017, 93, 220–233. [Google Scholar] [CrossRef] [Green Version]
- Shah, L.; Yahya, M.; Shah, S.M.A.; Nadeem, M.; Ali, A.; Ali, A.; Wang, J.; Riaz, M.W.; Rehman, S.; Wu, W.; et al. Improving Lodging Resistance: Using Wheat and Rice as Classical Examples. Int. J. Mol. Sci. 2019, 20, 4211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, T.; Ishimaru, K. Identification and Functional Analysis of a Locus for Improvement of Lodging Resistance in Rice. Plant Physiol. 2004, 134, 676–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashiwagi, T.; Madoka, Y.; Hirotsu, N.; Ishimaru, K. Locus prl5 improves lodging resistance of rice by delaying senescence and increasing carbohydrate reaccumulation. Plant Physiol. Biochem. 2006, 44, 152–157. [Google Scholar] [CrossRef]
- Yano, K.; Ookawa, T.; Aya, K.; Ochiai, Y.; Hirasawa, T.; Ebitani, T.; Takarada, T.; Yano, M.; Yamamoto, T.; Fukuoka, S.; et al. Isolation of a Novel Lodging Resistance QTL Gene Involved in Strigolactone Signaling and Its Pyramiding with a QTL Gene Involved in Another Mechanism. Mol. Plant 2015, 8, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Fan, C.; Li, Y.; Hu, Z.; Hu, H.; Wang, G.; Li, A.; Wang, Y.; Tu, Y.; Xia, T.; Peng, L.; et al. Ectopic expression of a novelOsExtensin-like gene consistently enhances plant lodging resistance by regulating cell elongation and cell wall thickening in rice. Plant Biotechnol. J. 2018, 16, 254–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sowadan, O.; Li, D.; Zhang, Y.; Zhu, S.; Hu, X.; Bhanbhro, L.B.; Edzesi, W.M.; Dang, X.; Hong, D. Mining of favorable alleles for lodging resistance traits in rice (oryza sativa) through association mapping. Planta 2018, 248, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.; Silk, W.K.; Schurr, U. Environmental Effects on Spatial and Temporal Patterns of Leaf and Root Growth. Annu. Rev. Plant Biol. 2009, 60, 279–304. [Google Scholar] [CrossRef]
- Ookawa, T.; Hobo, T.; Yano, M.; Murata, K.; Ando, T.; Miura, H.; Asano, K.; Ochiai, Y.; Ikeda, M.; Nishitani, R.; et al. New approach for rice improvement using a pleiotropic QTL gene for lodging resistance and yield. Nat. Commun. 2010, 1, 132. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Qian, Q.; Zhou, Y.; Yan, M.; Sun, L.; Zhang, M.; Fu, Z.; Wang, Y.; Han, B.; Pang, X.; et al. BRITTLE CULM1, Which Encodes a COBRA-Like Protein, Affects the Mechanical Properties of Rice Plants. Plant Cell 2003, 15, 2020–2031. [Google Scholar] [CrossRef] [Green Version]
- Rao, Y.; Yang, Y.; Xu, J.; Li, X.; Leng, Y.; Dai, L.; Huang, L.; Shao, G.; Ren, D.; Hu, J.; et al. EARLY SE-NESCENCE1 Encodes a SCAR-LIKE PROTEIN2 That Affects Water Loss in Rice. Plant Physiol. 2015, 169, 1225–1239. [Google Scholar] [CrossRef] [Green Version]
- Kong, X.; Gao, X.; Sun, W.; An, J.; Zhao, Y.; Zhang, H. Cloning and functional characterization of a cation–chloride co-transporter gene OsCCC1. Plant Mol. Biol. 2011, 75, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.C.; Yamaji, N.; Fujii-Kashino, M.; Ma, J.F. A Cation-Chloride Cotransporter Gene Is Required for Cell Elongation and Osmoregulation in Rice. Plant Physiol. 2016, 171, 494–507. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Wang, Y.; Long, Q.; Huang, J.; Wang, Y.; Zhou, K.; Zheng, M.; Sun, J.; Chen, H.; Chen, S.; et al. Overexpression of OsZHD1, a zinc finger homeodomain class homeobox transcription factor, induces abaxially curled and drooping leaf in rice. Planta 2014, 239, 803–816. [Google Scholar] [CrossRef]
- Wu, R.; Li, S.; He, S.; Waßmann, F.; Yu, C.; Qin, G.; Schreiber, L.; Qu, L.-J.; Gu, H. CFL1, a WW Domain Protein, Regulates Cuticle Development by Modulating the Function of HDG1, a Class IV Homeodomain Transcription Factor, in Rice and Arabidopsis. Plant Cell 2011, 23, 3392–3411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Luo, J.; Shao, G.; Wei, X.; Tang, S.; Sheng, Z.; Song, J.; Hu, P. Fine mapping of a major QTL for flag leaf width in rice, qFLW4, which might be caused by alternative splicing of NAL1. Plant Cell Rep. 2012, 31, 863–872. [Google Scholar] [CrossRef]
- Zhang, G.-H.; Li, S.-Y.; Wang, L.; Ye, W.-J.; Zeng, D.-L.; Rao, Y.-C.; Peng, Y.-L.; Hu, J.; Yang, Y.-L.; Xu, J.; et al. LSCHL4 from Japonica Cultivar, Which Is Allelic to NAL1, Increases Yield of Indica Super Rice 93-11. Mol. Plant 2014, 7, 1350–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shoujiang, Y.; Fang, J.; Lou, L.; Zhao, J.; Yuan, S.; Yin, L.; Sun, W.; Peng, L.; Guo, B.; Lixiang, P. Characterization of a Null Allelic Mutant of the Rice NAL1 Gene Reveals Its Role in Regulating Cell Division. PLoS ONE 2015, 10, e0118169. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Zhao, Y.; Liu, F.; Chen, Q.; Qi, J. Narrow leaf 1 (NAL1) regulates leaf shape by affecting cell expansion in rice (Oryza sativa L.). Biochem. Biophys. Res. Commun. 2019, 516, 957–962. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Guo, M.; Xu, L.; Wang, X.; Zhao, H.; Wang, J.; Yi, K. An SPX-RLI1 Module Regulates Leaf Inclination in Response to Phosphate Availability in Rice. Plant Cell 2018, 30, 853–870. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Zhang, X.; He, B.; Diao, L.; Sheng, S.; Wang, J.; Guo, X.; Su, N.; Wang, L.; Jiang, L.; et al. A Chlorophyll-Deficient Rice Mutant with Impaired Chlorophyllide Esterification in Chlorophyll Biosynthesis. Plant Physiol. 2007, 145, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Shi, Z.; Wang, J.; Wan, X.; Shen, G.; Wang, X.; Zhang, J. Over-expression of rice OsAGO7 gene induces upward curling of the leaf blade that enhanced erect-leaf habit. Planta 2007, 226, 99–108. [Google Scholar] [CrossRef]
- Li, L.; Shi, Z.-Y.; Shen, G.-Z.; Wang, X.-Q.; An, L.-S.; Zhang, J.-L. Overexpression of ACL1 (abaxially curled leaf 1) Increased Bulliform Cells and Induced Abaxial Curling of Leaf Blades in Rice. Mol. Plant 2010, 3, 807–817. [Google Scholar] [CrossRef]
- Wang, Z.-K.; Huang, Y.-X.; Miao, Z.-D.; Hu, Z.-Y.; Song, X.-Z.; Liu, L. Identification and characterization of BGL11(t), a novel gene regulating leaf-color mutation in rice (Oryza sativa L.). Genes Genom. 2013, 35, 491–499. [Google Scholar] [CrossRef]
- Lin, Y.; Tan, L.; Zhao, L.; Sun, X.; Sun, C. RLS3, a protein with AAA+ domain localized in chloroplast, sustains leaf longevity in rice. J. Integr. Plant Biol. 2016, 58, 971–982. [Google Scholar] [CrossRef]
- Yang, S.-Q.; Li, W.-Q.; Miao, H.; Gan, P.-F.; Qiao, L.; Chang, Y.-L.; Shi, C.-H.; Chen, K.-M. REL2, A Gene Encoding An Unknown Function Protein which Contains DUF630 and DUF632 Domains Controls Leaf Rolling in Rice. Rice 2016, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- The 3000 rice genomes project. GigaScience 2014, 3, 2047–2217X.
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Depristo, M.A.; Banks, E.; Poplin, R.; Garimella, K.V.; Maguire, J.R.; Hartl, C.; Philippakis, A.A.; Del Angel, G.; Rivas, M.A.; Hanna, M.; et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011, 43, 491–498. [Google Scholar] [CrossRef]
- Howie, B.N.; Donnelly, P.; Marchini, J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [Green Version]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel- accelerated tool for Genome-Wide Associ-ation Study. bioRxiv 2020. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Wei, X.; Sang, T.; Zhao, Q.; Feng, Q.; Zhao, Y.; Li, C.; Zhu, C.; Lu, T.; Zhang, Z.; et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 2010, 42, 961–967. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Liu, X.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Xie, W.; Gong, L.; Lu, K.; Wang, W.; Li, Y.; Liu, X.; Zhang, H.; Dong, H.; et al. Genome-wide association analyses provide genetic and biochemical insights into natural variation in rice metabolism. Nat. Genet. 2014, 46, 714–721. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, W.; Xing, H.; Yan, J.; Meng, X.; Li, X.; Fu, X.; Xu, J.; Lian, X.; Yu, S.; et al. Genetic Architecture of Natural Variation in Rice Chlorophyll Content Revealed by a Genome-Wide Association Study. Mol. Plant 2015, 8, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Guo, Z.; Yang, W.; Chang, Y.; Ma, X.; Tu, H.; Xiong, F.; Jiang, N.; Feng, H.; Huang, C.; Yang, P.; et al. Genome-Wide Association Studies of Image Traits Reveal Genetic Architecture of Drought Re-sistance in Rice. Mol. Plant 2018, 11, 789–805. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Pang, Y.; Wang, C.; Chen, K.; Zhu, Y.; Shen, C.; Ali, J.; Xu, J.; Li, Z. New Candidate Genes Affecting Rice Grain Appearance and Milling Quality Detected by Genome-Wide and Gene-Based Association Analyses. Front. Plant Sci. 2017, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, K.; Yamamoto, E.; Aya, K.; Takeuchi, H.; Lo, P.-C.; Hu, L.; Yamasaki, M.; Yoshida, S.; Kitano, H.; Hirano, K.; et al. Genome-wide association study using whole-genome sequencing rapidly identifies new genes influencing agronomic traits in rice. Nat. Genet. 2016, 48, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Zhu, X.; Wang, Q.; Zhang, J.; Chen, H.; Dong, G.; Zhu, L.; Zheng, H.; Xie, Q.; Nian, J.; et al. RiceTUTOU1 Encodes a Suppressor of cAMP Receptor-Like Protein That Is Important for Actin Organization and Panicle Development. Plant Physiol. 2015, 169, 1179–1191. [Google Scholar] [CrossRef] [Green Version]
- Fekih, R.; Tamiru, M.; Kanzaki, H.; Abe, A.; Yoshida, K.; Kanzaki, E.; Saitoh, H.; Takagi, H.; Natsume, S.; Undan, J.R.; et al. The rice (Oryza sativa L.) LESION MIMIC RESEMBLING, which encodes an AAA-type ATPase, is implicated in defense response. Mol. Genet. Genom. 2015, 290, 611–622. [Google Scholar] [CrossRef]
- Zhu, X.; Yin, J.; Liang, S.; Liang, R.; Zhou, X.; Chen, Z.; Zhao, W.; Wang, J.; Li, W.; He, M.; et al. The Multivesicular Bodies (MVBs)-Localized AAA ATPase LRD6-6 Inhibits Immunity and Cell Death Likely through Regulating MVBs-Mediated Vesic-ular Trafficking in Rice. PLoS Genet. 2016, 12, e1006311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khush, G.S. Origin, dispersal, cultivation and variation of rice. Plant Mol. Biol. 1997, 35, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lübberstedt, T. Molecular basis of trait correlations. Trends Plant Sci. 2010, 15, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Liu, X.; Zhang, B.; Yuan, X.; Xing, Y.; Liu, H.; Luo, L.; Chen, G.; Xiong, L. Genetic analyses of lodging resistance and yield provide insights into post-Green-Revolution breeding in rice. Plant Biotechnol. J. 2021, 19, 814–829. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Shimono, M.; Maeda, S.; Inoue, H.; Mori, M.; Hasegawa, M.; Sugano, S.; Takatsuji, H. Suppression of the Rice Fatty-Acid Desaturase GeneOsSSI2 Enhances Resistance to Blast and Leaf Blight Diseases in Rice. Mol. Plant-Microbe Interact. 2009, 22, 820–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Marker No. | Size (Mb) | Spacing (kb) |
|---|---|---|---|
| chr1 | 457,835 | 43.2 | 0.094 |
| chr2 | 387,459 | 35.9 | 0.092 |
| chr3 | 361,044 | 36.3 | 0.101 |
| chr4 | 368,017 | 35.5 | 0.096 |
| chr5 | 280,079 | 29.7 | 0.106 |
| chr6 | 333,313 | 31.1 | 0.093 |
| chr7 | 306,057 | 29.7 | 0.097 |
| chr8 | 335,328 | 28.4 | 0.085 |
| chr9 | 260,134 | 22.9 | 0.088 |
| chr10 | 282,524 | 23.1 | 0.082 |
| chr11 | 380,739 | 29.0 | 0.076 |
| chr12 | 324,308 | 29.4 | 0.091 |
| Total | 4,076,837 | 372.2 | 0.091 |
| QTL | Year | CHROM | POS | REF | ALT | Effect | SE | p |
|---|---|---|---|---|---|---|---|---|
| qLFR2.1 | 2019 | 2 | 4,243,076 | G | A | 0.06 | 0.011 | 1.17 × 10−7 |
| 2020 | 2 | 4,461,960 | G | T | 0.05 | 0.010 | 4.24 × 10−8 | |
| qLFR2.3 | 2019 | 2 | 18,545,189 | A | G | 0.03 | 0.005 | 1.82 × 10−7 |
| qLFR5.3 | 2020 | 5 | 20,540,653 | A | G | 0.06 | 0.011 | 1.42 × 10−8 |
| 2019 | 5 | 20,540,653 | A | G | 0.05 | 0.009 | 4.64 × 10−8 | |
| qLFR9.1 | 2019 | 9 | 17,764,668 | C | A | 0.04 | 0.006 | 3.06 × 10−8 |
| qSID8.1 | 2020 | 8 | 8,673,481 | G | A | 0.28 | 0.050 | 4.97 × 10−8 |
| 2019 | 8 | 8,722,341 | C | T | 0.35 | 0.053 | 1.32 × 10−10 | |
| qIL1.1 | 2019 | 1 | 5,920,879 | C | T | 1.85 | 0.341 | 8.34 × 10−8 |
| qIL1.2 | 2020 | 1 | 32,933,806 | G | T | 1.65 | 0.299 | 6.10 × 10−8 |
| 2019 | 1 | 32,934,166 | T | C | 1.61 | 0.300 | 1.39 × 10−7 | |
| qIL8.1 | 2019 | 8 | 8,396,436 | C | T | 1.50 | 0.283 | 2.32 × 10−7 |
| 2020 | 8 | 8,717,396 | C | T | 1.75 | 0.334 | 1.84 × 10−7 | |
| qIL8.2 | 2020 | 8 | 14,215,369 | G | A | 1.70 | 0.304 | 4.22 × 10−8 |
| qSPAD7 | 2019 | 7 | 5,119,605 | G | A | −1.12 | 0.212 | 1.92 × 10−7 |
| qSPAD11.1 | 2020 | 11 | 27,695,597 | C | A | −1.54 | 0.284 | 1.00 × 10−7 |
| qSWT4.3 | 2019 | 4 | 29,983,581 | T | A | −0.03 | 0.008 | 1.59 × 10−7 |
| 2020 | 4 | 30,459,506 | A | G | −0.06 | 0.010 | 9.45 × 10−8 | |
| qSWT5.2 | 2020 | 5 | 6,243,695 | C | T | 0.02 | 0.004 | 1.27 × 10−7 |
| 2019 | 5 | 6,245,355 | C | T | 0.01 | 0.003 | 1.97 × 10−8 | |
| qSWT7.3 | 2019 | 7 | 28,576,217 | T | C | −0.07 | 0.013 | 2.06 × 10−7 |
| 2020 | 7 | 28,576,224 | T | C | −0.07 | 0.013 | 2.06 × 10−7 | |
| qFLW8 | 2020 | 8 | 22,563,133 | T | C | 0.22 | 0.041 | 8.67 × 10−8 |
| 2019 | 8 | 22,849,319 | C | T | 0.21 | 0.038 | 4.24 × 10−8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, B.; Wang, T.; Luo, Y.; Xu, D.; Li, L.; Diao, Y.; Gao, Z.; Hu, Z.; Zheng, X. Genome-Wide Association Study Identified Novel Candidate Loci/Genes Affecting Lodging Resistance in Rice. Genes 2021, 12, 718. https://doi.org/10.3390/genes12050718
Meng B, Wang T, Luo Y, Xu D, Li L, Diao Y, Gao Z, Hu Z, Zheng X. Genome-Wide Association Study Identified Novel Candidate Loci/Genes Affecting Lodging Resistance in Rice. Genes. 2021; 12(5):718. https://doi.org/10.3390/genes12050718
Chicago/Turabian StyleMeng, Bingxin, Tao Wang, Yi Luo, Deze Xu, Lanzhi Li, Ying Diao, Zhiyong Gao, Zhongli Hu, and Xingfei Zheng. 2021. "Genome-Wide Association Study Identified Novel Candidate Loci/Genes Affecting Lodging Resistance in Rice" Genes 12, no. 5: 718. https://doi.org/10.3390/genes12050718
APA StyleMeng, B., Wang, T., Luo, Y., Xu, D., Li, L., Diao, Y., Gao, Z., Hu, Z., & Zheng, X. (2021). Genome-Wide Association Study Identified Novel Candidate Loci/Genes Affecting Lodging Resistance in Rice. Genes, 12(5), 718. https://doi.org/10.3390/genes12050718