
Differential mRNA and miRNA Profiles Reveal the Potential Roles of Genes and miRNAs Involved in LPS Infection in Chicken Macrophages

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture and LPS Stimulation
2.2. RNA Extraction
2.3. The mRNA Library and Small RNA Preparation and Sequencing
2.4. Differential Expression Analysis and Functional Enrichment Analysis
2.5. Real-Time Quantitative PCR of mRNA and miRNAs
3. Results
3.1. Transcriptome Library Construction
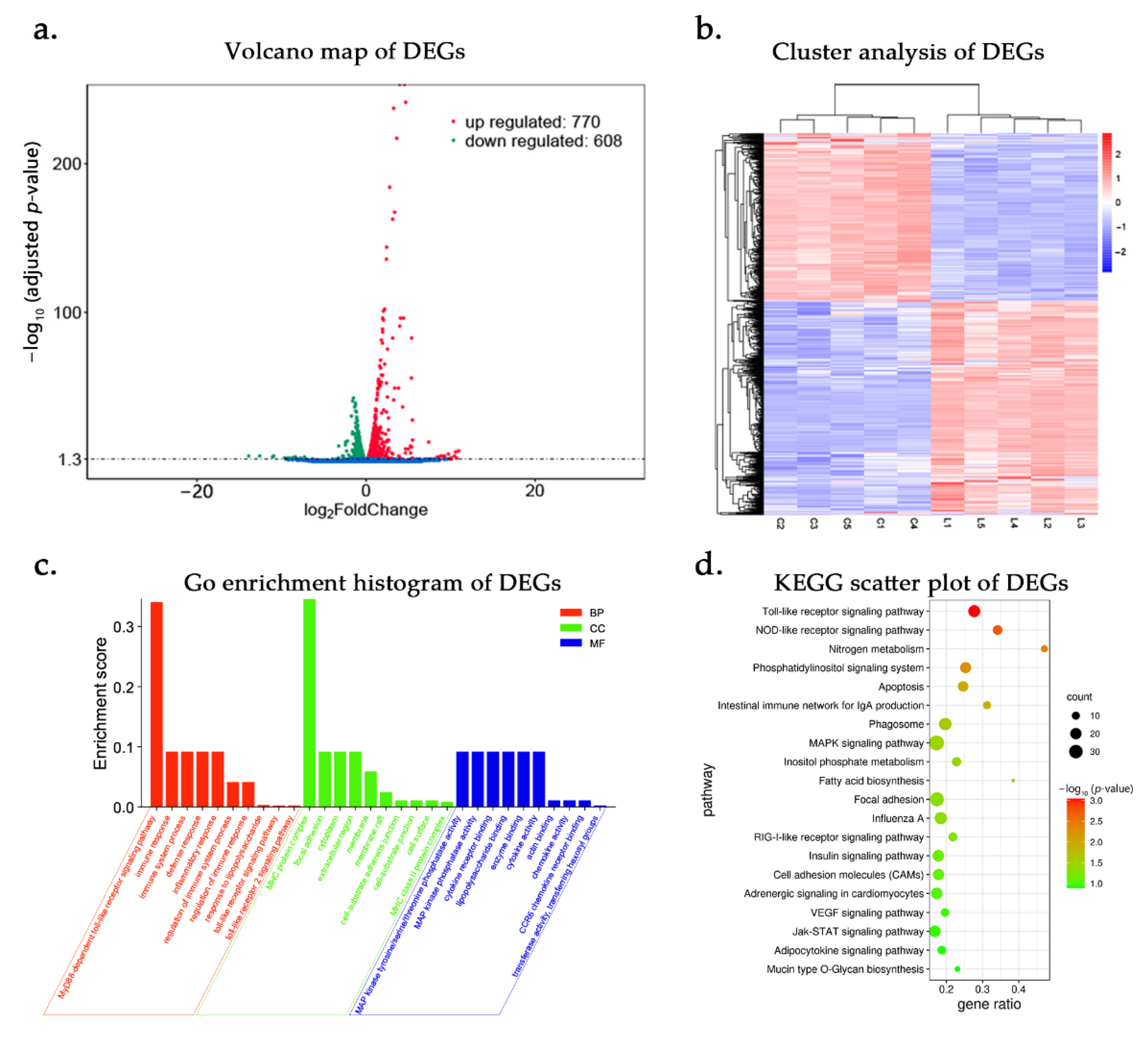
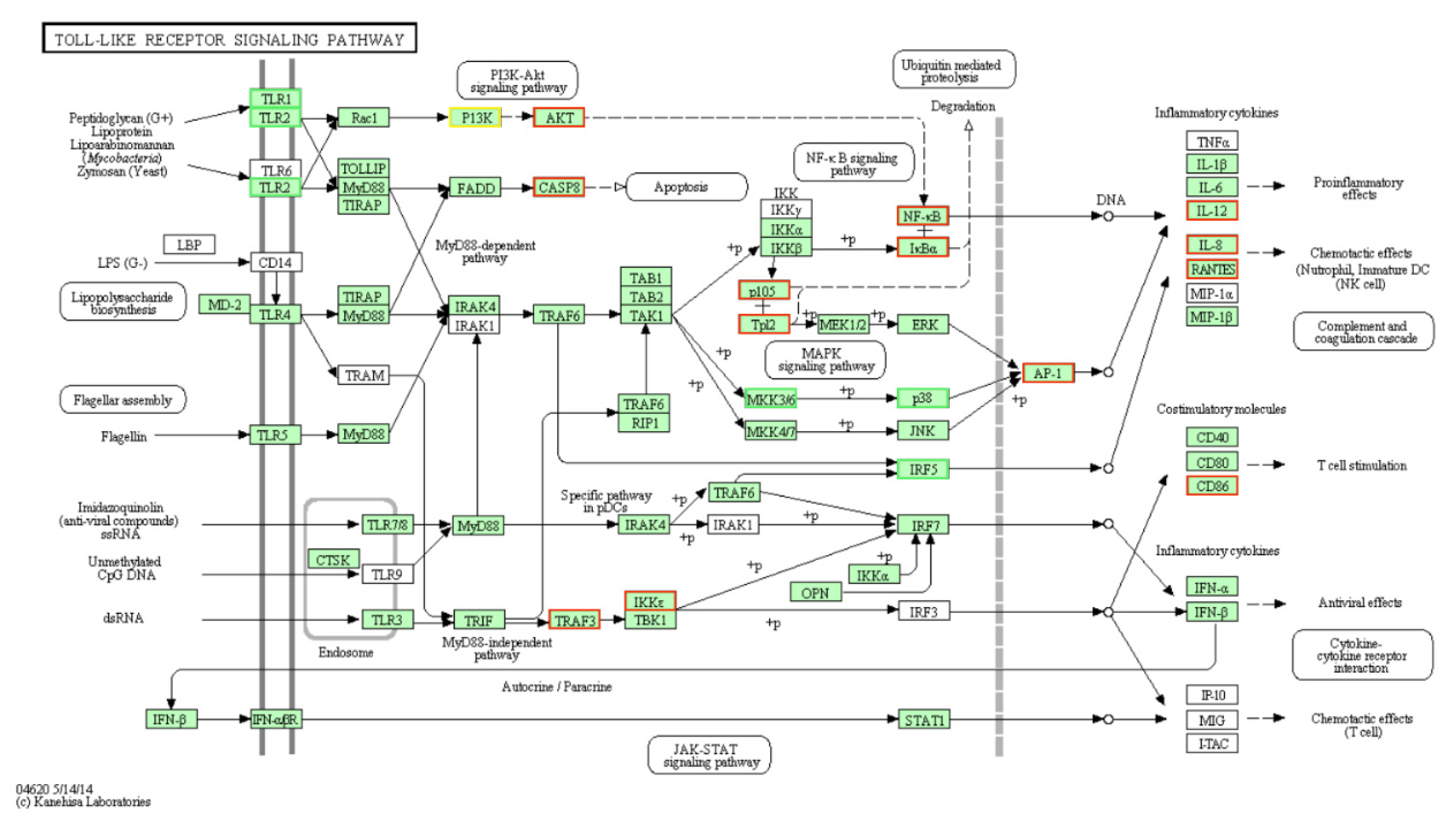
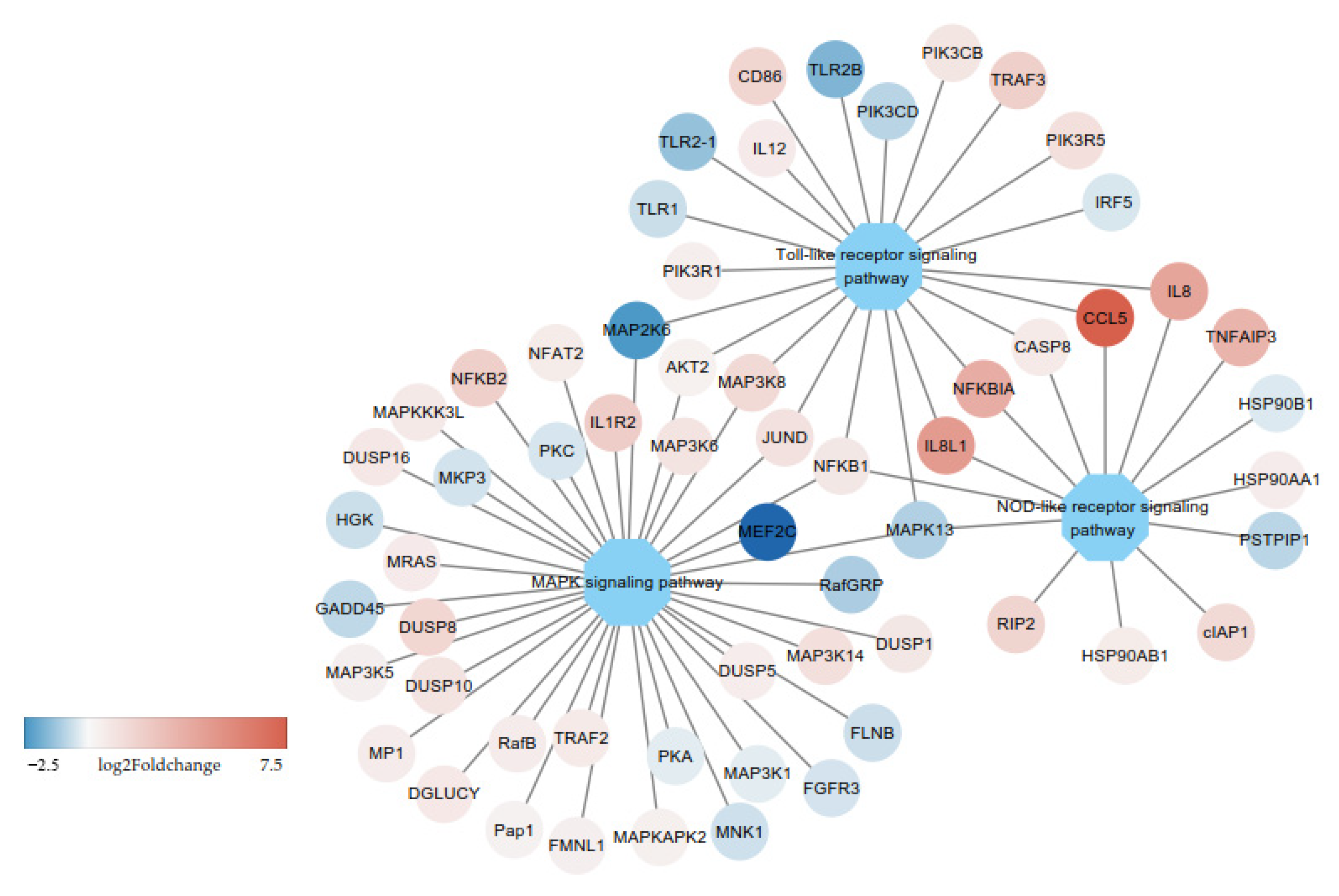
3.2. DE-mRNAs and Functional Annotation
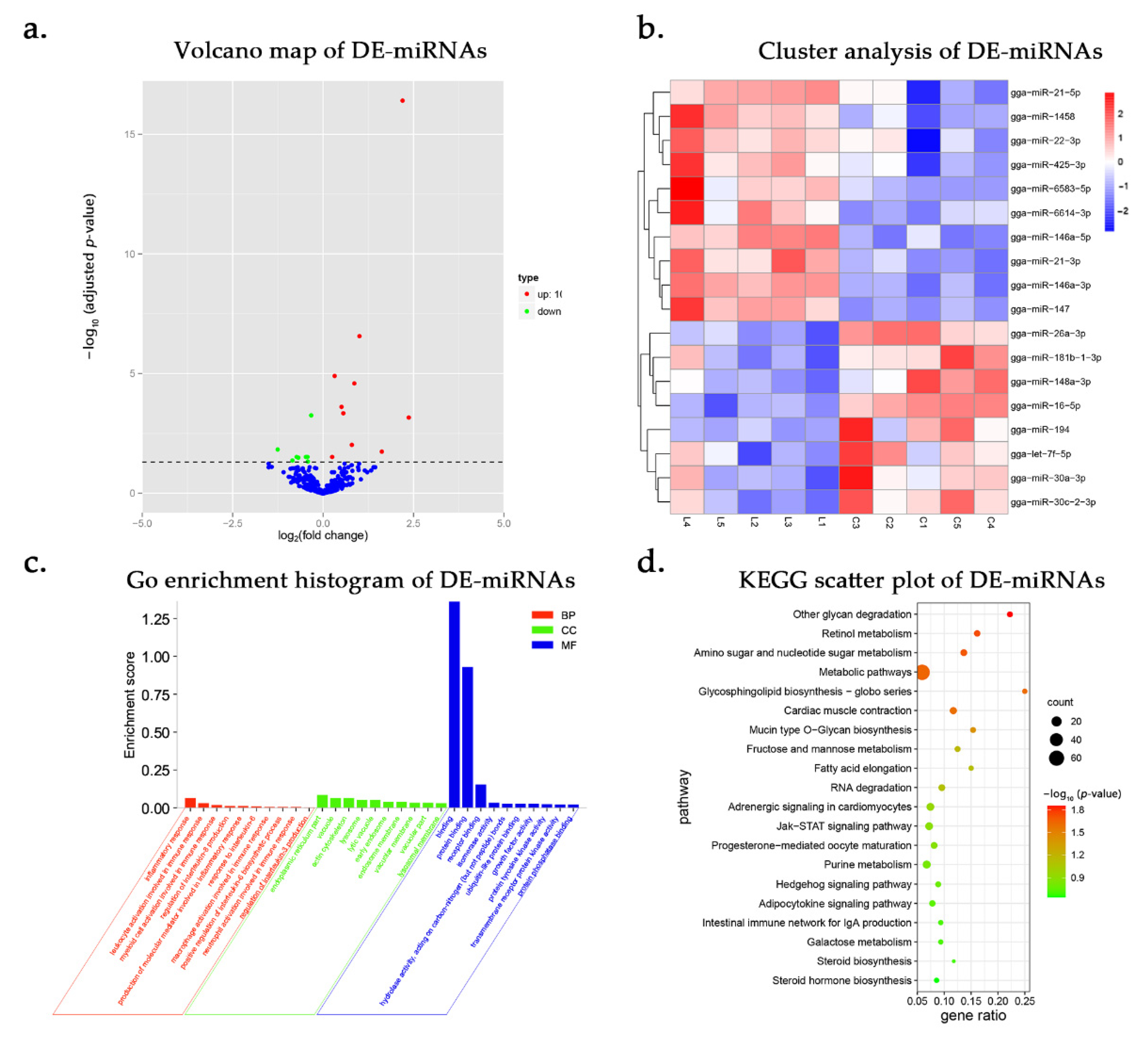
3.3. DE-miRNAs and Functional Annotation
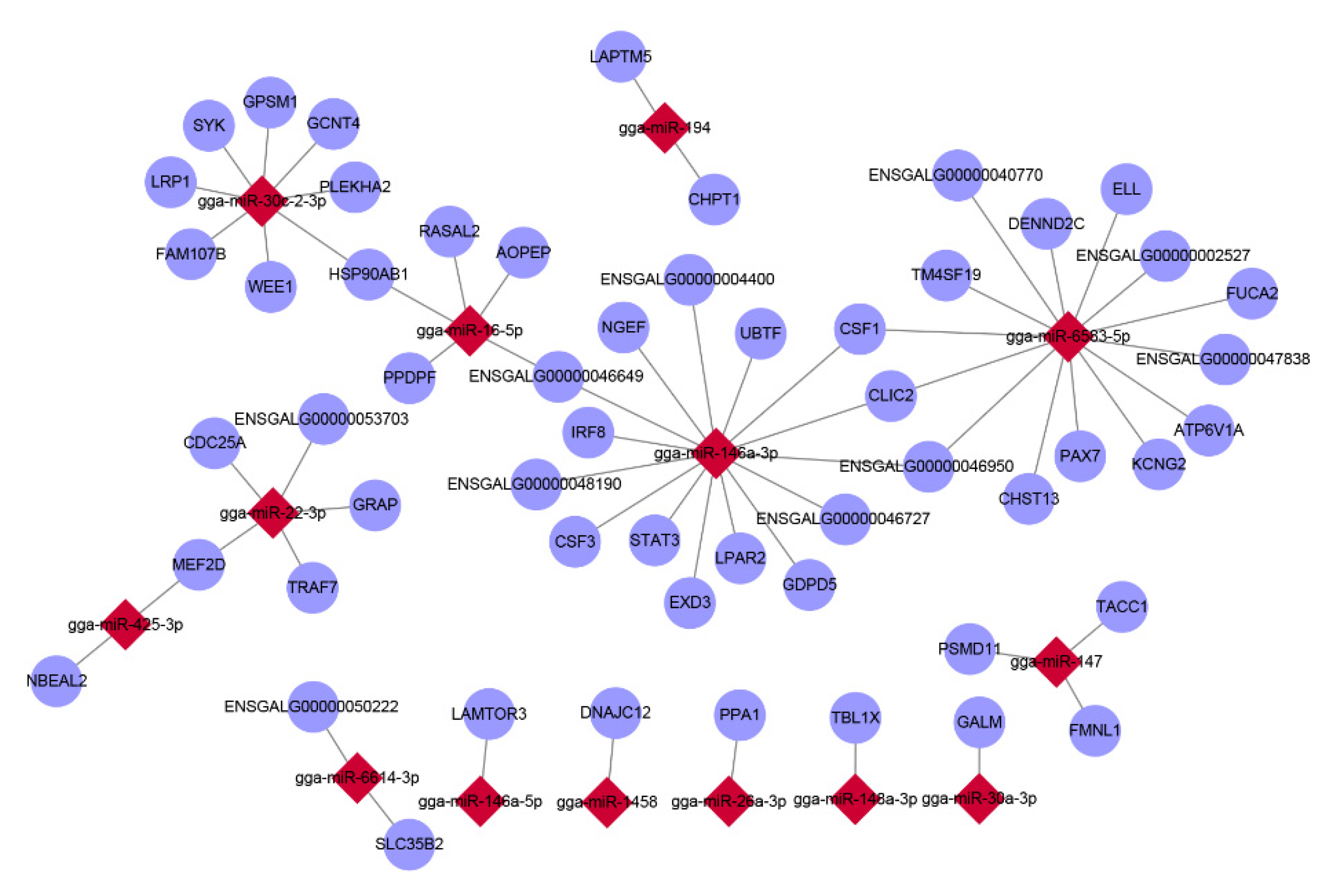
3.4. Correlation Analysis of mRNA and miRNA
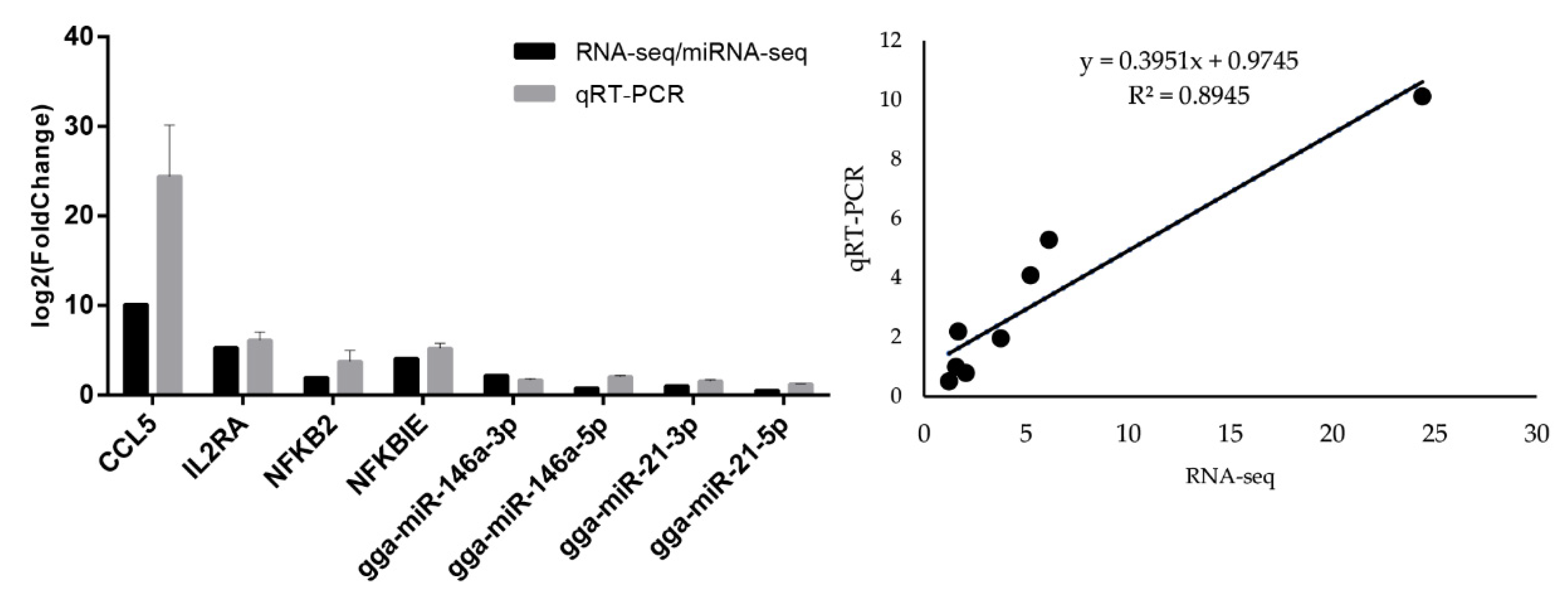
3.5. Quantitative Real-Time PCR of mRNAs and miRNAs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kellenberger, E.; Ryter, A. Cell Wall and Cytoplasmic Membrane of Escherichia coli. J. Cell Biol. 1958, 4, 323–326. [Google Scholar] [CrossRef]
- Bladen, H.A.; Mergenhagen, S.E. Ultrastructure of Veillonella and morphological correlation of an outer membrane with particles associated with endotoxic activity. J. Bacteriol. 1964, 88, 1482–1492. [Google Scholar] [CrossRef]
- Galanos, C.; Lüderitz, O.; Rietschel, E.T.; Westphal, O.; Brade, H.; Brade, L.; Freudenberg, M.; Schade, U.; Imoto, M.; Yoshimura, H.; et al. Synthetic and natural Escherichia coli free lipid A express identical endotoxic activities. JBIC J. Biol. Inorg. Chem. 1985, 148, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, A.A.; Hellerqvist, C.G. Bacteriophage Attachment Sites, Serological Specificity, and Chemical Composition of the Lipopolysaccharides of Semirough and Rough Mutants of Salmonella typhimurium. J. Bacteriol. 1971, 105, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Wright, S.D.; A Ramos, R.; Tobias, P.S.; Ulevitch, R.J.; Mathison, J.C. CD14, a receptor for complexes of lipopolysaccharide (LPS) and LPS binding protein. Science 1990, 249, 1431–1433. [Google Scholar] [CrossRef]
- Fujiwara, N. Macrophages in Inflammation. Curr. Drug Target Inflamm. Allergy 2005, 4, 281–286. [Google Scholar] [CrossRef]
- White, C.R. Allometric Analysis beyond Heterogeneous Regression Slopes: Use of the Johnson-Neyman Technique in Comparative Biology. Physiol. Biochem. Zool. 2003, 76, 135–140. [Google Scholar] [CrossRef][Green Version]
- Jin, W.; Guangtian, Y. Role of nuclear transcription factor-κB in endotoxin-induced shock in rats. Acta Acad. Med. Wuhan 2005, 25, 174–177. [Google Scholar] [CrossRef]
- Aderka, D.; Le, J.M.; Vilcek, J. IL-6 inhibits lipopolysaccharide-induced tumor necrosis factor production in cultured human monocytes, U937 cells, and in mice. J. Immunol. 1989, 143, 3517–3523. [Google Scholar]
- Segreti, J.; Gheusi, G.; Dantzer, R.; Kelley, K.W.; Johnson, R.W. Defect in Interleukin-1β Secretion Prevents Sickness Behavior in C3H/HeJ Mice. Physiol. Behav. 1997, 61, 873–878. [Google Scholar] [CrossRef]
- Kawasaki, H.; Taira, K. Functional analysis of microRNAs during the retinoic acid-induced neuronal differentiation of human NT2 cells. Nucleic Acids Symp. Ser. 2003, 3, 243–244. [Google Scholar] [CrossRef] [PubMed]
- Brennecke, J.; Hipfner, D.R.; Stark, A.; Russell, R.B.; Cohen, S.M. bantam Encodes a Developmentally Regulated microRNA that Controls Cell Proliferation and Regulates the Proapoptotic Gene hid in Drosophila. Cell 2003, 113, 25–36. [Google Scholar] [CrossRef]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro-RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.P.; Burge, C.B.; Bartel, D.P. Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes are MicroRNA Targets. Cell 2005, 120, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Yan, W.; Wang, S.E.; Baltimore, D. Dual mechanisms of posttranscriptional regulation of Tet2 by Let-7 microRNA in macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 12416–12421. [Google Scholar] [CrossRef]
- Li, X.; Huang, S.; Yu, T.; Liang, G.; Liu, H.; Pu, D.; Peng, N. MiR-140 modulates the inflammatory responses of Mycobacterium tuberculosis -infected macrophages by targeting TRAF6. J. Cell. Mol. Med. 2019, 23, 5642–5653. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, W.; Li, Q.; Wang, J.; Liu, R.; Everaert, N.; Liu, J.; Zhang, Y.; Zheng, M.; Cui, H.; et al. Splenic microRNA Expression Profiles and Integration Analyses Involved in Host Responses to Salmonella enteritidis Infection in Chickens. Front. Cell. Infect. Microbiol. 2017, 7, 377. [Google Scholar] [CrossRef]
- Sun, W.; Liu, R.; Li, P.; Li, Q.; Cui, H.; Zheng, M.; Wen, J.; Zhao, G. Chicken gga-miR-1306-5p targets Tollip and plays an important role in host response against Salmonella enteritidis infection. J. Anim. Sci. Biotechnol. 2019, 10, 59. [Google Scholar] [CrossRef]
- Friedländer, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2011, 40, 37–52. [Google Scholar] [CrossRef]
- Wen, M.; Shen, Y.; Shi, S.; Tang, T. miREvo: An integrative microRNA evolutionary analysis platform for next-generation sequencing experiments. BMC Bioinform. 2012, 13, 140. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, F.; Wang, Q.; Zhang, N.; Zheng, J.; Zheng, M.; Liu, R.; Cui, H.; Wen, J.; Zhao, G. SPOP promotes ubiquitination and degradation of MyD88 to suppress the innate immune response. PLoS Pathog. 2020, 16, e1008188. [Google Scholar] [CrossRef] [PubMed]
- Withanage, G.S.K.; Kaiser, P.; Wigley, P.; Powers, C.; Mastroeni, P.; Brooks, H.; Barrow, P.; Smith, A.; Maskell, D.; McConnell, I. Rapid Expression of Chemokines and Proinflammatory Cytokines in Newly Hatched Chickens Infected with Salmonella enterica Serovar Typhimurium. Infect. Immun. 2004, 72, 2152–2159. [Google Scholar] [CrossRef]
- Kim, Y.K.; Shin, J.-S.; Nahm, M.H. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. Toll-like Receptors and Their Crosstalk with Other Innate Receptors in Infection and Immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.-R.; Ge, J.; Li, W.-Y.; Zhou, W.-C.; Xu, L.; Geng, D.-Q. NF-κB/let-7f-5p/IL-10 pathway involves in wear particle-induced osteolysis by inducing M1 macrophage polarization. Cell Cycle 2018, 17, 2134–2145. [Google Scholar] [CrossRef] [PubMed]
- Gysler, S.M.; Mulla, M.J.; Guerra, M.; Brosens, J.J.; Salmon, J.E.; Chamley, L.W.; Abrahams, V.M. Antiphospholipid antibody-induced miR-146a-3p drives trophoblast interleukin-8 secretion through activation of Toll-like receptor 8. Mol. Hum. Reprod. 2016, 22, 465–474. [Google Scholar] [CrossRef]
- Tang, C.-T.; Liang, Q.; Yang, L.; Lin, X.-L.; Wu, S.; Chen, Y.; Zhang, X.-T.; Gao, Y.-J.; Ge, Z.-Z. RAB31 Targeted by MiR-30c-2-3p Regulates the GLI1 Signaling Pathway, Affecting Gastric Cancer Cell Proliferation and Apoptosis. Front. Oncol. 2018, 8, 554. [Google Scholar] [CrossRef]
- Karki, R.; Lee, E.; Place, D.; Samir, P.; Mavuluri, J.; Sharma, B.R.; Balakrishnan, A.; Malireddi, R.S.; Geiger, R.; Zhu, Q.; et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 2018, 173, 920–933.e13. [Google Scholar] [CrossRef]
- Karki, R.; Lee, E.; Sharma, B.R.; Banoth, B.; Kanneganti, T.-D. IRF8 Regulates Gram-Negative Bacteria–Mediated NLRP3 Inflammasome Activation and Cell Death. J. Immunol. 2020, 204, 2514–2522. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.-J.; Hu, S.-J.; Zhao, Q.-Q.; Liu, X.-S.; Liu, C.; Wang, H. Toll-like receptor 4 (TLR4) deficiency aggravates dextran sulfate sodium (DSS)-induced intestinal injury by down-regulating IL6, CCL2 and CSF3. Ann. Transl. Med. 2019, 7, 713. [Google Scholar] [CrossRef] [PubMed]
- Temperley, N.D.; Berlin, S.; Paton, I.R.; Griffin, D.K.; Burt, D.W. Evolution of the chicken Toll-like receptor gene family: A story of gene gain and gene loss. BMC Genom. 2008, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 2004, 432, 695–716. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Li, X.; Qu, G.; Wang, Y.; Li, Q.; Guo, Y.; Hou, L.; Liu, J.; Eko, F.O.; He, C. Correction: Chlamydia psittaci PmpD-N Modulated Chicken Macrophage Function by Triggering Th2 Polarization and the TLR2/MyD88/NF-κB Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 2639. [Google Scholar] [CrossRef]
- Meurer, R.; Van Riper, G.; Feeney, W.; Cunningham, P.; Hora, D.; Springer, M.S.; E Macintyre, D.; Rosen, H. Formation of eosinophilic and monocytic intradermal inflammatory sites in the dog by injection of human RANTES but not human monocyte chemoattractant protein 1, human macrophage inflammatory protein 1 alpha, or human interleukin 8. J. Exp. Med. 1993, 178, 1913–1921. [Google Scholar] [CrossRef]
- Xu, Z.; Yoshida, T.; Wu, L.; Maiti, D.; Cebotaru, L.; Duh, E.J. Transcription Factor MEF2C Suppresses Endothelial Cell Inflammation via Regulation of NF-κB and KLF2. J. Cell. Physiol. 2015, 230, 1310–1320. [Google Scholar] [CrossRef]
- Zhang, X.; Guo, Y.; Xu, X.; Tang, T.; Sun, L.; Wang, H.; Zhou, W.; Fang, L.; Li, Q.; Xie, P. miR-146a promotes Borna disease virus 1 replication through IRAK1/TRAF6/NF-κB signaling pathway. Virus Res. 2019, 271, 197671. [Google Scholar] [CrossRef]
- Li, Y.; Zhu, H.; Wei, X.; Li, H.; Yu, Z.; Zhang, H.; Liu, W. LPS induces HUVEC angiogenesis in vitro through miR-146a-mediated TGF-beta1 inhibition. Am. J. Transl. Res. 2017, 9, 591–600. [Google Scholar]
- Liu, J.; Ma, Z.; Ran, Z. MiR-21-3p modulates lipopolysaccharide-induced inflammation and apoptosis via targeting TGS 4 in retinal pigment epithelial cells. Clin. Exp. Pharmacol. Physiol. 2019, 46, 883–889. [Google Scholar] [CrossRef]
- Amura, T.; Yanai, H.; Savitsky, D.; Taniguchi, T. The IRF Family Transcription Factors in Immunity and Oncogenesis. Annu. Rev. Immunol. 2008, 26, 535–584. [Google Scholar] [CrossRef]
- Salem, S.; Salem, D.; Gros, P. Role of IRF8 in immune cells functions, protection against infections, and susceptibility to inflammatory diseases. Qual. Life Res. 2020, 139, 707–721. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Zhao, X.; Huo, R.; Xu, T. IRF3 and IRF8 Regulate NF-κB Signaling by Targeting MyD88 in Teleost Fish. Front. Immunol. 2020, 11, 606. [Google Scholar] [CrossRef] [PubMed]
- Salem, S.; Gros, P. Genetic Determinants of Susceptibility to Mycobacterial Infections: IRF8, A New Kid on the Block. Adv. Exp. Med. Biol. 2013, 783, 45–80. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Wang, J.; Zhang, J.; Wen, J.; Zhao, G.; Li, Q. Differential mRNA and miRNA Profiles Reveal the Potential Roles of Genes and miRNAs Involved in LPS Infection in Chicken Macrophages. Genes 2021, 12, 760. https://doi.org/10.3390/genes12050760
Zhang Q, Wang J, Zhang J, Wen J, Zhao G, Li Q. Differential mRNA and miRNA Profiles Reveal the Potential Roles of Genes and miRNAs Involved in LPS Infection in Chicken Macrophages. Genes. 2021; 12(5):760. https://doi.org/10.3390/genes12050760
Chicago/Turabian StyleZhang, Qi, Jie Wang, Jin Zhang, Jie Wen, Guiping Zhao, and Qinghe Li. 2021. "Differential mRNA and miRNA Profiles Reveal the Potential Roles of Genes and miRNAs Involved in LPS Infection in Chicken Macrophages" Genes 12, no. 5: 760. https://doi.org/10.3390/genes12050760
APA StyleZhang, Q., Wang, J., Zhang, J., Wen, J., Zhao, G., & Li, Q. (2021). Differential mRNA and miRNA Profiles Reveal the Potential Roles of Genes and miRNAs Involved in LPS Infection in Chicken Macrophages. Genes, 12(5), 760. https://doi.org/10.3390/genes12050760