
Population Genetics of the Invasive Red Fox, Vulpes vulpes, in South-Eastern Australia



Abstract
:1. Introduction
2. Methods
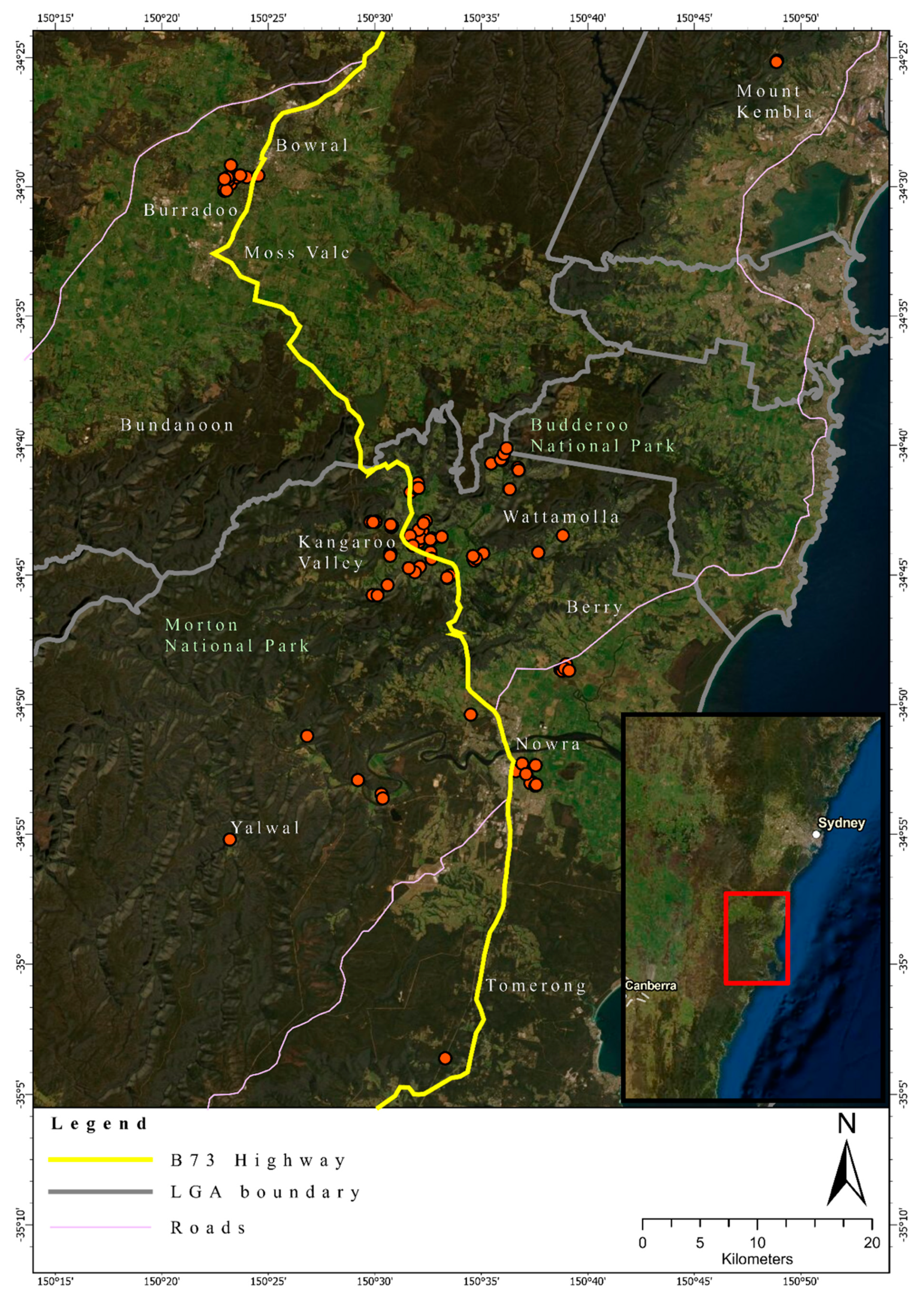
2.1. Study Location and Tissue Collection
2.2. DNA Extractions
2.3. Genotyping
2.4. Quality Control and Data Analysis
3. Results
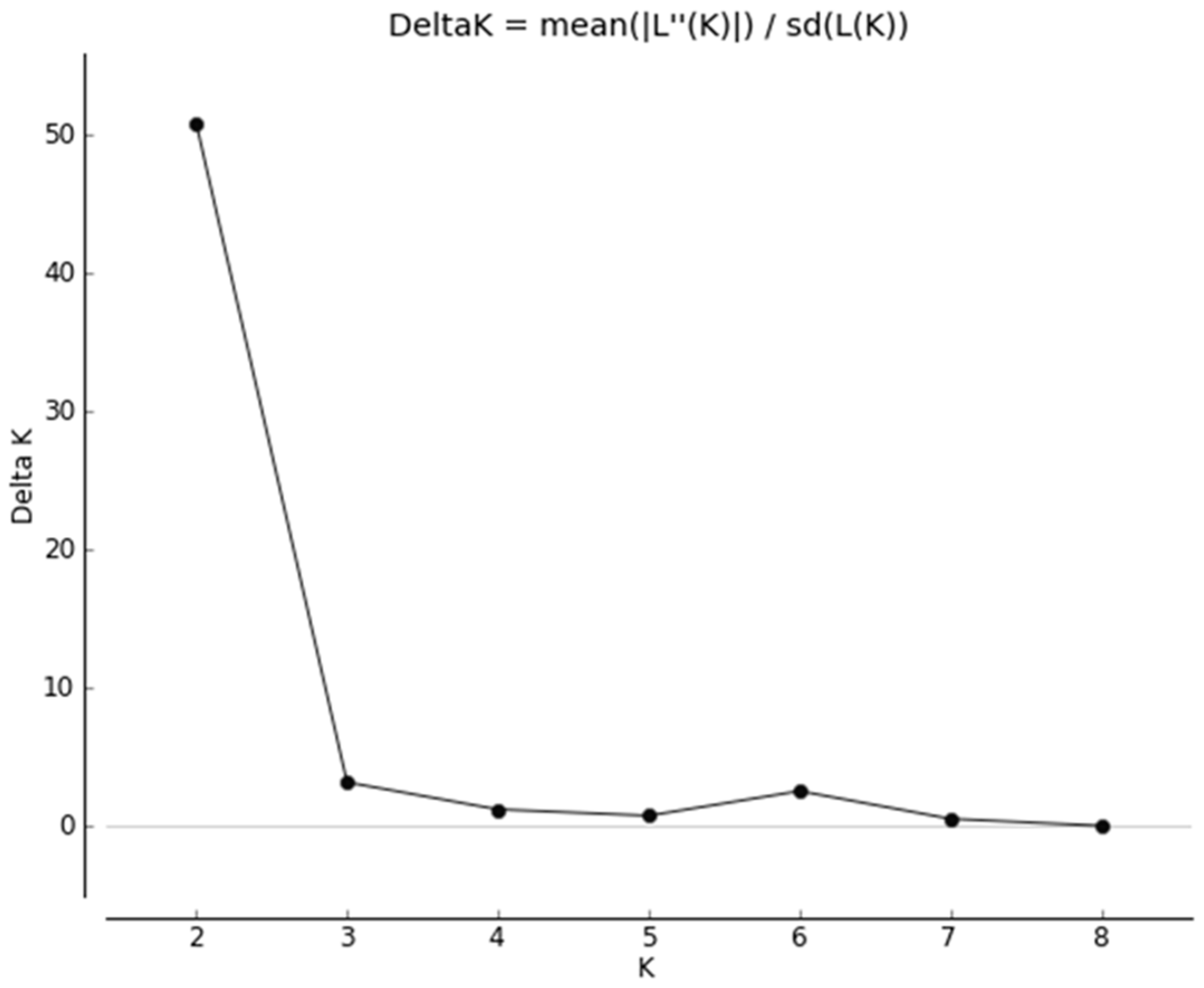
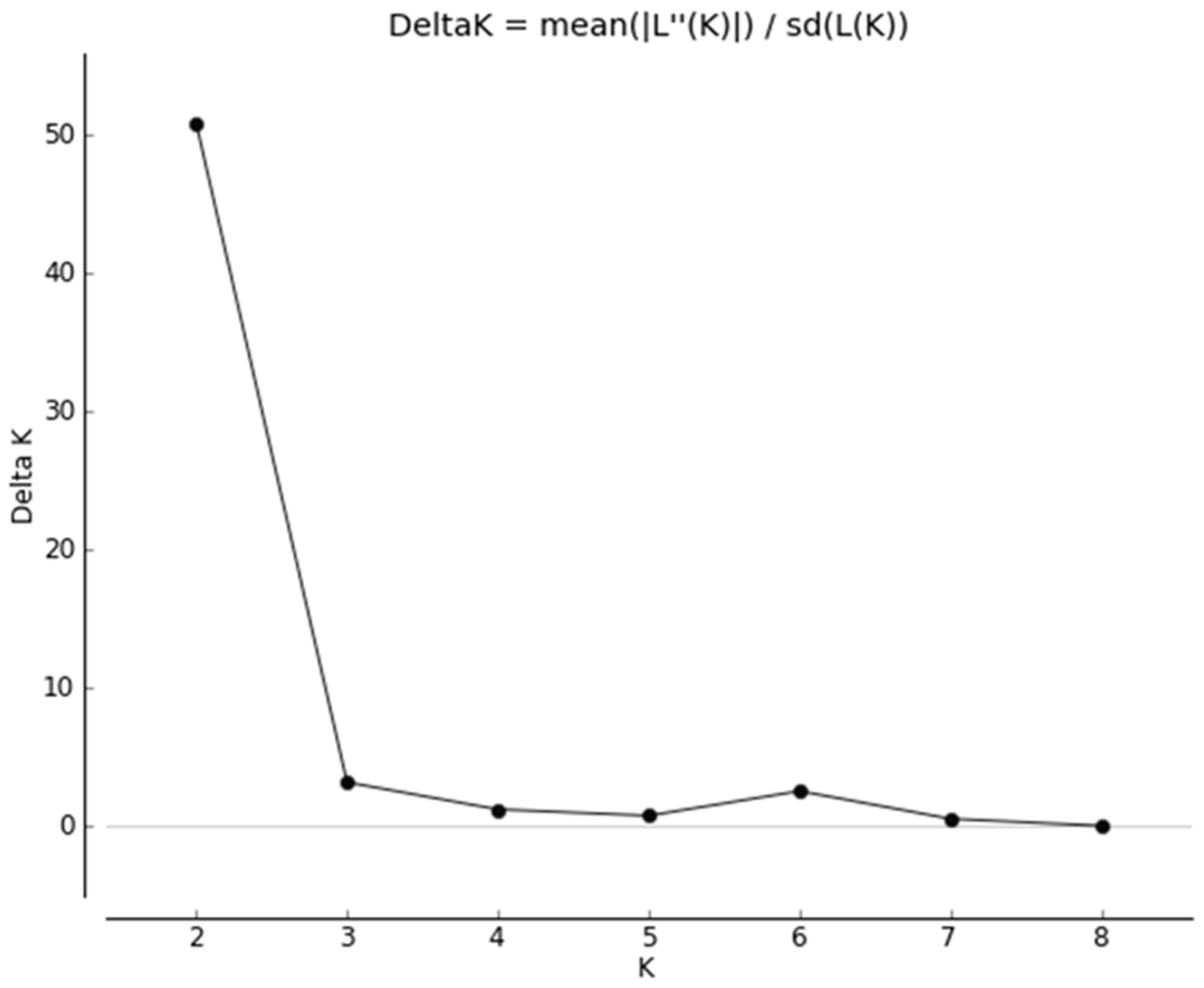
3.1. Quality Control
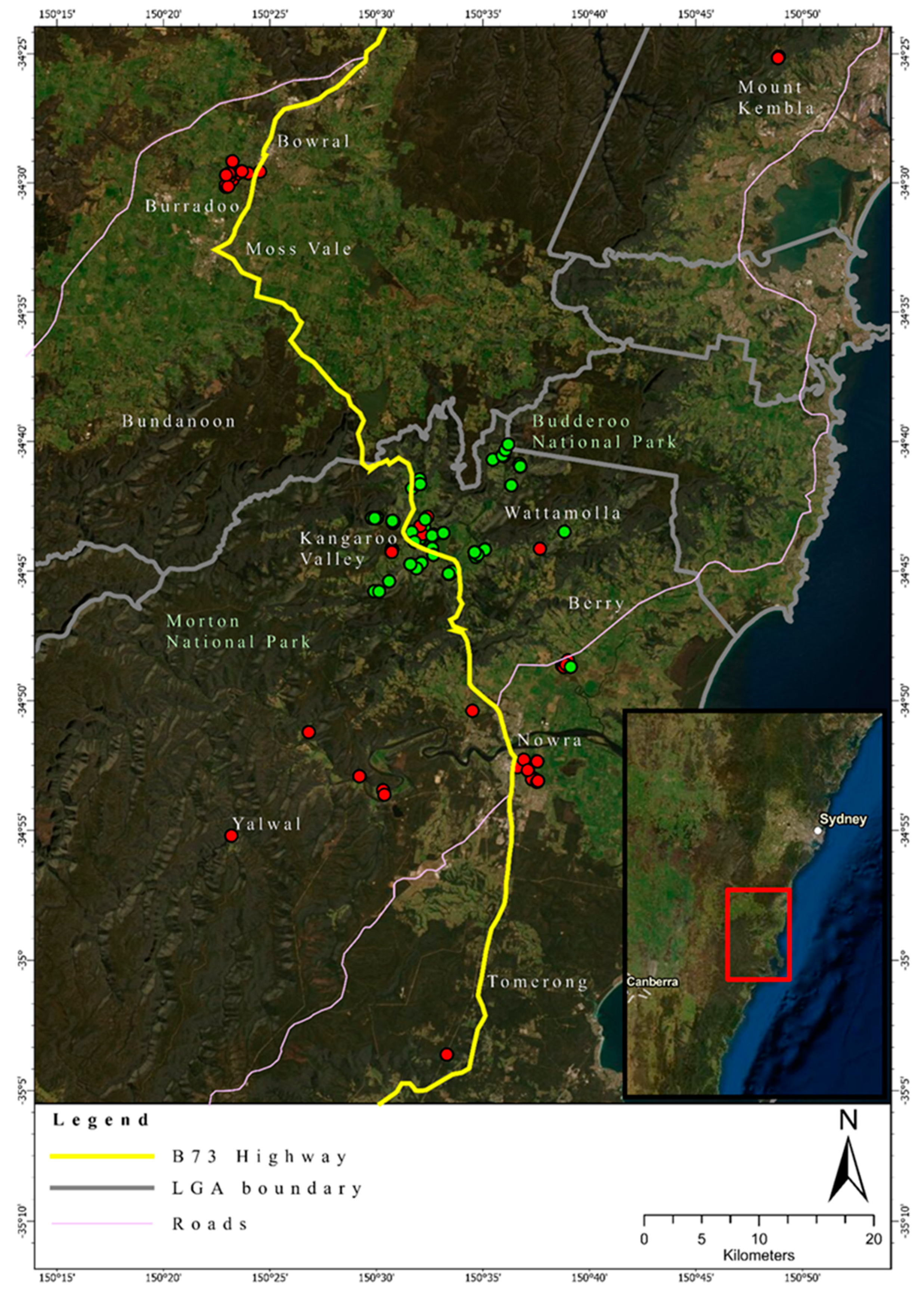
3.2. Heterozygosity of the One- and Two-Cluster Models
3.3. Relatedness
3.4. Sex-Biased Dispersal
4. Discussion
5. Management Implications and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fairfax, R.J. Dispersal of the introduced red fox (Vulpes vulpes) across Australia. Biol. Invasions 2018, 21, 1259–1268. [Google Scholar] [CrossRef]
- Edwards, C.; Soulsbury, C.; Statham, M.; Ho, S.; Wall, D.; Dolf, G.; Iossa, G.; Baker, P.; Harris, S.; Sacks, B.; et al. Temporal genetic variation of the red fox, Vulpes vulpes, across western Europe and the British Isles. Quat. Sci. Rev. 2012, 57, 95–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, P. Assessing invasive animals in Australia 2008. In Invasive Animals Cooperative Research Centre; National Land & Water Resources Audit: Canberra, Australia, 2008. [Google Scholar]
- Dickman, C.R. Impact of exotic generalist predators on the native fauna of Australia. Wildl. Biol. 1996, 2, 185–195. [Google Scholar] [CrossRef]
- Kinnear, J.; Sumner, N.; Onus, M. The red fox in Australia—An exotic predator turned biocontrol agent. Biol. Conserv. 2002, 108, 335–359. [Google Scholar] [CrossRef]
- Woinarski, J.C.Z.; Burbidge, A.A.; Harrison, P.L. Ongoing unraveling of a continental fauna: Decline and extinction of Australian mammals since European settlement. Proc. Natl. Acad. Sci. USA 2015, 112, 4531–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisher, D.O.; Johnson, C.N.; Lawes, M.J.; Fritz, S.A.; McCallum, H.; Blomberg, S.P.; VanDerWal, J.; Abbott, B.; Frank, A.; Legge, S.; et al. The current decline of tropical marsupials in Australia: Is history repeating? Glob. Ecol. Biogeogr. 2013, 23, 181–190. [Google Scholar] [CrossRef]
- Johnson, C.N.; Isaac, J.L. Body mass and extinction risk in Australian marsupials: The ‘Critical Weight Range’ revisited. Austral Ecol. 2009, 34, 35–40. [Google Scholar] [CrossRef]
- Augee, M.; Smith, B.; Rose, S. Survival of Wild and Hand-reared Ringtail Possums (Pseudocheirus peregrinus) in Bushland near Sydney. Wildl. Res. 1996, 23, 99–108. [Google Scholar] [CrossRef]
- Finlayson, G.; Finlayson, S.; Dickman, C. Returning the rat-kangaroos: Translocation attempts in the family Potoroidae (Superfamily Macropodoidea) and recommendations for conservation. In Macropods: The Biology of Kangaroos, Wallabies and Rat-Kangaroos; Coulson, G., Eldridge, M., Eds.; CSIRO Publishing: Melbourne, Australia, 2010; pp. 245–262. [Google Scholar]
- Dufty, A.; Seebeck, J.; McKay, J.; Watson, A. Reintroduction of the eastern barred bandicoot Perameles gunnii at Gellibrand Hill Park, Victoria. In Reintroduction Biology of Australian and New Zealand Fauna; Serena, M., Ed.; Surrey Beatty & Sons: Sydney, Australia, 1994; pp. 219–225. [Google Scholar]
- Pietsch, R. The fate of urban common brushtail possums translocated to sclerophyll forest. In Reintroduction Biology of Australian and New Zealand Fauna; Serena, M., Ed.; Surrey Beatty & Sons: Sydney, Australia, 1994; pp. 236–246. [Google Scholar]
- Glen, A.S.; Dickman, C.R. Home range, denning behaviour and microhabitat use of the carnivorous marsupial Dasyurus maculatus in eastern Australia. J. Zool. 2006, 268, 347–354. [Google Scholar] [CrossRef]
- Glen, A.S.; Dickman, C.R. Niche overlap between marsupial and eutherian carnivores: Does competition threaten the endangered spotted-tailed quoll? J. Appl. Ecol. 2008, 45, 700–707. [Google Scholar] [CrossRef]
- Saunders, G.R.; Gentle, M.N.; Dickman, C.R. The impacts and management of foxes Vulpes vulpes in Australia. Mammal Rev. 2010, 40, 181–211. [Google Scholar] [CrossRef]
- Robertson, B.C.; Gemmell, N.J. Defining eradication units to control invasive pests. J. Appl. Ecol. 2004, 41, 1042–1048. [Google Scholar] [CrossRef]
- Abdelkrim, J.; Pascal, M.; Calmet, C.; Samadi, S. Importance of Assessing Population Genetic Structure before Eradication of Invasive Species: Examples from Insular Norway Rat Populations. Conserv. Biol. 2005, 19, 1509–1518. [Google Scholar] [CrossRef]
- Rollins, L.A.; Woolnough, A.P.; Wilton, A.N.; Sinclair, R.; Sherwin, W.B. Invasive species can’t cover their tracks: Using microsatellites to assist management of starling (Sturnus vulgaris) populations in Western Australia. Mol. Ecol. 2009, 18, 1560–1573. [Google Scholar] [CrossRef] [PubMed]
- Veale, A.J.; Edge, K.-A.; McMurtrie, P.; Fewster, R.M.; Clout, M.N.; Gleeson, D.M. Using genetic techniques to quantify reinvasion, survival and in situ breeding rates during control operations. Mol. Ecol. 2013, 22, 5071–5083. [Google Scholar] [CrossRef] [PubMed]
- Atterby, H.; Allnutt, T.R.; MacNicoll, A.D.; Jones, E.P.; Smith, G.C. Population genetic structure of the red fox (Vulpes vulpes) in the UK. Mammal Res. 2014, 60, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Zecchin, B.; De Nardi, M.; Nouvellet, P.; Vernesi, C.; Babbucci, M.; Crestanello, B.; Bagó, Z.; Bedeković, T.; Hostnik, P.; Milani, A.; et al. Genetic and spatial characterization of the red fox (Vulpes vulpes) population in the area stretching between the Eastern and Dinaric Alps and its relationship with rabies and canine distemper dynamics. PLoS ONE 2019, 14, e0213515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morin, P.; Luikart, G.; Wayne, R.; SNP workshop group. SNPs in ecology, evolution and conservation. Trends Ecol. Evol. 2004, 19, 208–216. [Google Scholar] [CrossRef]
- Puckett, E.E.; Park, J.; Combs, M.; Blum, M.J.; Bryant, J.E.; Caccone, A.; Costa, F.; Deinum, E.E.; Esther, A.; Himsworth, C.G.; et al. Global population divergence and admixture of the brown rat (Rattus norvegicus ). Proc. R. Soc. B Boil. Sci. 2016, 283, 20161762. [Google Scholar] [CrossRef] [Green Version]
- McCann, B.E.; Smyser, T.J.; Schmit, B.; Newman, R.A.; Piaggio, A.J.; Malek, M.J.; Swafford, S.R.; Sweitzer, R.A.; Sweitzer, R.A.; Simmons, R.B. Molecular population structure for feral swine in the United States: Population Structure for Feral Swine. J. Wildl. Manag. 2018, 82, 821–832. [Google Scholar] [CrossRef] [Green Version]
- Nørgaard, L.S.; Mikkelsen, D.M.G.; Elmeros, M.; Chriél, M.; Madsen, A.B.; Nielsen, J.L.; Pertoldi, C.; Randi, E.; Fickel, J.; Brygida, S.; et al. Population genomics of the raccoon dog (Nyctereutes procyonoides) in Denmark: Insights into invasion history and population development. Biol. Invasions 2017, 19, 1637–1652. [Google Scholar] [CrossRef]
- Helyar, S.J.; Hemmer-Hansen, J.; Bekkevold, D.; Taylor, M.I.; Ogden, R.; Limborg, M.T.; Cariani, A.; Maes, G.E.; Diopere, E.; Carvalho, G.R.; et al. Application of SNPs for population genetics of nonmodel organisms: New opportunities and challenges. Mol. Ecol. Resour. 2011, 11, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Kukekova, A.V.; Johnson, J.L.; Xiang, X.; Feng, S.; Liu, S.; Rando, H.M.; Kharlamova, A.V.; Herbeck, Y.; Serdyukova, N.A.; Xiong, Z.; et al. Red fox genome assembly identifies genomic regions associated with tame and aggressive behaviours. Nat. Ecol. Evol. 2018, 2, 1479–1491. [Google Scholar] [CrossRef] [PubMed]
- Shoalhaven Fox Control Project: Landholders and Volunteers Working Together to Control Foxes in the Shoalhaven. Available online: https://landcare.nsw.gov.au/groups/shoalhaven-landcare-association-inc/shoalhaven-fox-control/ (accessed on 5 July 2020).
- Kilian, A.; Wenzl, P.; Huttner, E.; Carling, J.; Xia, L.; Blois, H.; Caig, V.; Heller-Uszynska, K.; Jaccoud, D.; Hopper, C.; et al. Diversity Arrays Technology: A Generic Genome Profiling Technology on Open Platforms. In Methods in Molecular Biology; Springer Science and Business Media LLC; Humana Press: Totowa, NJ, USA, 2012; Volume 888, pp. 67–89. [Google Scholar]
- Melville, J.; Haines, M.L.; Boysen, K.; Hodkinson, L.; Kilian, A.; Date, K.L.S.; Potvin, D.A.; Parris, K.M. Identifying hybridization and admixture using SNPs: Application of the DArTseq platform in phylogeographic research on vertebrates. R. Soc. Open Sci. 2017, 4, 161061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCBI Vulpes Vulpes Annotation Release 100. Available online: https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Vulpes_vulpes/100/ (accessed on 27 May 2020).
- Jombart, T.; Ahmed, I. Adegenet 1.3-1: New tools for the analysis of genome-wide SNP data. Bioinformatics 2011, 27, 3070–3071. [Google Scholar] [CrossRef] [Green Version]
- Gruber, B.; Unmack, P.; Berry, O.F.; Georges, A. Dartr: An r package to facilitate analysis of SNP data generated from reduced representation genome sequencing. Mol. Ecol. Resour. 2018, 18, 691–699. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.R-project.org/ (accessed on 27 May 2020).
- Goudet, J. Hierfstat, a package for r to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.; Daly, M.; et al. PLINK: A toolset for whole-genome association and population-based linkage analysis. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2011, 4, 359–361. [Google Scholar] [CrossRef]
- Goudet, J.; Perrin, N.; Waser, P. Tests for sex-biased dispersal using bi-parentally inherited genetic markers. Mol. Ecol. 2002, 11, 1103–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lariviere, S.; Pasitschniak-Arts, M. Vulpes vulpes. Mamm. Species 1996, 537, 1–11. [Google Scholar] [CrossRef]
- Saunders, I.W.; Brohede, J.; Hannan, G.N. Estimating genotyping error rates from Mendelian errors in SNP array genotypes and their impact on inference. Genomics 2007, 90, 291–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walton, Z.; Hagenlund, M.; Østbye, K.; Samelius, G.; Odden, M.; Norman, A.; Willebrand, T.; Spong, G. Moving far, staying close: Red fox dispersal patterns revealed by SNP genotyping. Conserv. Genet. 2021, 22, 249–257. [Google Scholar] [CrossRef]
- Norén, K.; Angerbjörn, A.; Wallén, J.; Meijer, T.; Sacks, B. Red foxes colonizing the tundra: Genetic analysis as a tool for population management. Conserv. Genet. 2017, 18, 359–370. [Google Scholar] [CrossRef] [Green Version]
- Austerlitz, F.; Jung-Muller, B.; Godelle, B.; Gouyon, P.-H. Evolution of Coalescence Times, Genetic Diversity and Structure during Colonization. Theor. Popul. Biol. 1997, 51, 148–164. [Google Scholar] [CrossRef] [Green Version]
- Sacks, B.N.; Statham, M.J.; Perrine, J.D.; Wisely, S.M.; Aubry, K.B. North American montane red foxes: Expansion, fragmentation, and the origin of the Sacramento Valley red fox. Conserv. Genet. 2010, 11, 1523–1539. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.C.; Cowan, P.E.; Fricke, B.L.; Geddes, S.; Hansen, B.D.; Lam, M.; Cooper, D.W. High microsatellite diversity and differential structuring among populations of the introduced common brushtail possum, Trichosurus vulpecula, in New Zealand. Genet. Res. 2004, 83, 101–111. [Google Scholar] [CrossRef]
- Campbell, C.D.; Sarre, S.D.; Stojanovic, D.; Gruber, B.; Medlock, K.; Harris, S.; MacDonald, A.J.; Holleley, C.E. When is a native species invasive? Incursion of a novel predatory marsupial detected using molecular and historical data. Divers. Distrib. 2018, 24, 831–840. [Google Scholar] [CrossRef] [Green Version]
- Spencer, P.B.S.; Giustiniano, D.; Hampton, J.O.; Gee, P.; Burrows, N.; Rose, K.; Martin, G.R.; Woolnough, A.P. Identification and management of a single large population of wild dromedary camels. J. Wildl. Manag. 2012, 76, 1254–1263. [Google Scholar] [CrossRef]
- Towerton, A.L.; Penman, T.D.; Kavanagh, R.P.; Dickman, C.R. Detecting pest and prey responses to fox control across the landscape using remote cameras. Wildl. Res. 2011, 38, 208–220. [Google Scholar] [CrossRef]
- Carter, A. Improving Red Fox (Vulpes vulpes) management for Bush Stone-curlew (Burhinus grallarius) conservation in south-eastern Australia. Ph.D. Thesis, Charles Sturt University, Australia, 2010. [Google Scholar]
- Towerton, A.L.; Kavanagh, R.P.; Penman, T.D.; Dickman, C.R. Ranging behaviour and movements of the red fox in remnant forest habitats. Wildl. Res. 2016, 43, 492–506. [Google Scholar] [CrossRef]
- Walton, Z.; Samelius, G.; Odden, M.; Willebrand, T. Long-distance dispersal in red foxes Vulpes vulpes revealed by GPS tracking. Eur. J. Wildl. Res. 2018, 64, 64. [Google Scholar] [CrossRef] [Green Version]
- Robinson, N.A.; Marks, C.A. Genetic structure and dispersal of red foxes (Vulpes vulpes) in urban Melbourne. Aust. J. Zool. 2001, 49, 589. [Google Scholar] [CrossRef]
- Schwartz, M.K.; Luikart, G.; Waples, R.S. Genetic monitoring as a promising tool for conservation and management. Trends Ecol. Evol. 2007, 22, 25–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rollins, L.A.; Woolnough, A.P.; Sherwin, W.B. Population genetic tools for pest management: A review. Wildl. Res. 2006, 33, 251–261. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genetic Cluster | N | HO | HE | FIS | FST |
|---|---|---|---|---|---|
| Cluster 1 | 42 | 0.297 | 0.311 | 0.041 (−0.283 to 0.300) | 0.018 |
| Cluster 2 | 51 | 0.290 | 0.309 | 0.057 (−0.018 to 0.193) |
| Pedigree | FoxID (Estimated Age) | IBS | Cluster Origin According to Structure | Type of Relationship | No. of Mendel Errors (%) | |
|---|---|---|---|---|---|---|
| 1 | 16 (<1 year) 17 (<1 year) | 0.86 | 0.54 | 2 | offspring of same mating event | N/A |
| 2 | 30 (2–3 years) 31 (<1 year) | 0.86 | 0.52 | 1 | parent–offspring relation (dyad) | 147 (0.82) |
| 3a | 66 (>5 years) 68 (3–4 years) | 0.84 | 0.47 | 1 | parent–offspring relation (dyad) | 147 (0.82) |
| 3b | 66 (>5 years) 69 (4–5 years) | 0.84 | 0.44 | 1 | parent–offspring relation (dyad) | 139 (0.77) |
| 3c | 68 (3–4 years) 69 (4–5 years) | 0.87 | 0.53 | 1 | offspring of potentially two mating events in two breeding seasons | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watson, K.M.-A.; Mikac, K.M.; Schwab, S.G. Population Genetics of the Invasive Red Fox, Vulpes vulpes, in South-Eastern Australia. Genes 2021, 12, 786. https://doi.org/10.3390/genes12050786
Watson KM-A, Mikac KM, Schwab SG. Population Genetics of the Invasive Red Fox, Vulpes vulpes, in South-Eastern Australia. Genes. 2021; 12(5):786. https://doi.org/10.3390/genes12050786
Chicago/Turabian StyleWatson, Kalynda M.-A., Katarina M. Mikac, and Sibylle G. Schwab. 2021. "Population Genetics of the Invasive Red Fox, Vulpes vulpes, in South-Eastern Australia" Genes 12, no. 5: 786. https://doi.org/10.3390/genes12050786
APA StyleWatson, K. M. -A., Mikac, K. M., & Schwab, S. G. (2021). Population Genetics of the Invasive Red Fox, Vulpes vulpes, in South-Eastern Australia. Genes, 12(5), 786. https://doi.org/10.3390/genes12050786