
Genetic Variant c.245A>G (p.Asn82Ser) in GIPC3 Gene Is a Frequent Cause of Hereditary Nonsyndromic Sensorineural Hearing Loss in Chuvash Population

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Characteristics of Patients
2.3. Characteristics of Healthy Individuals
2.4. Molecular Genetic Methods
2.4.1. Sequencing of GJB2, GJB3, and GJB6 Genes
2.4.2. Multiplex Ligase-Dependent Probe Amplification (MLPA) Analysis
2.4.3. Whole Exome Sequencing
2.4.4. Sequencing of GIPC3 Gene Coding Region
2.4.5. Analysis for the NM_133261.3(GIPC3):c.245A>G (p.Asn82Ser) Variant in GIPC3 Gene
3. Results
4. Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Nucleotide Variant (Amino Acid Change) | Mutation Type | Domain | Number of Patients/Number of Families | Hearing Loss Severity | Population | Reference |
|---|---|---|---|---|---|---|---|
| 1 | NM_133261.3(GIPC3):c.122C>A (p.Thr41Lys) | Missense | GH1 | 1/1 | Severe to profound | Saudi Arabia | [33] |
| 2 | NM_133261.3(GIPC3):c.136G>A (p.Gly46Arg) | Missense | GH1 | 1/1 | Severe to profound | Pakistan | [32] |
| 3 | NM_133261.3(GIPC3):c.226-1G>T | Splicing site | NA1 | 1/1 | Severe to profound | Pakistan | [35] |
| 4 | NM_133261.3(GIPC3):c.245A>G (p.Asn82Ser) | Missense | GH1 | 1/1 | Severe | Iran | [39] |
| 2/1 | Moderate; moderate to severe | Iran | [40] | ||||
| 5 | NM_133261.3(GIPC3):c.264G>A (p.Met88Ile) | Missense | GH1 | 1/1 | Mild to severe | Pakistan | [32] |
| 6 | NM_133261.3(GIPC3):c.281G>A (p.Gly94Asp) | Missense | GH1 | 1/1 | Mild to severe | Pakistan | [32] |
| 7 | NM_133261.3(GIPC3):c.472G>A (p.Glu158Lys) | Missense | PDZ | 5/1 | Severe to profound | Iran (Arab origin) | [36] |
| 8 | NM_133261.3(GIPC3):c.508C>A (p.His170Asn) | Missense | PDZ | 1/1 | AR NSHL | Turkey | [34] |
| 1/1 | AR NSHL | Turkey | [35] | ||||
| 9 | NM_133261.3(GIPC3):c.565C>T (p.Arg189Cys) | Missense | PDZ | 1/1 | Severe to profound | Pakistan | [32] |
| 10 | NM_133261.3(GIPC3):c.662C>T (p.Thr221Ile) | Missense | GH2 | 1/1 | Profound | Pakistan | [32] |
| 11 | NM_133261.3(GIPC3):c.685dupG (p.Ala229GlyfsTer10) | Frameshift | H2 | 1/1 | Moderate to severe | Pakistan | [32] |
| 12 | NM_133261.3(GIPC3):c.767G>A (p.Gly256Asp) | Missense | GH2 | 1/1 | Moderate to severe | Pakistan | [32] |
| 13 | NM_133261.3(GIPC3):c.785T>G (p.Leu262Arg) | Missense | GH2 | 1/1 | Stable, profound | India | [31] |
| 14 | NM_133261.3(GIPC3):c.903G>A (p.Trp301Ter) | Nonsense | GH2 | 1/1 | Progressive, profound | Holland | [31] |
| 15 | NM_133261.3(GIPC3):c.759C>G (p.Ser253Arg) | Missense | GH2 | 1/1 | Severe | Pakistan | [37] |
| 16 | NM_133261.3(GIPC3):c.764T>A (p.Met255Lys) | Missense | GH2 | 2/1 | Severe to profound | Algeria | [38] |
| 17 | NM_133261.3(GIPC3):c.937T>C (p.Ter313GlnextTer98) | Stop-loss | GH2 | 5/1 | ND | Pakistan | [41] |
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Korver, A.M.; Smith, R.J.; Van Camp, G.; Schleiss, M.R.; Bitner-Glindzicz, M.A.; Lustig, L.R.; Usami, S.I.; Boudewyns, A.N. Congenital hearing loss. Nat. Rev. Dis. Primers 2017, 12, 16094. [Google Scholar] [CrossRef] [PubMed]
- Markova, T.G.; Bliznetz, E.A.; Polyakov, A.V.; Tavartkiladze, G.A. Twenty years of clinical studies of GJB2-linked hearing loss in Russia. Vestn. Otorinolaringol. 2018, 83, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Meena, R.; Ayub, M. Genetics of human hereditary hearing impairment. J. Ayub. Med. Coll. Abbottabad 2017, 29, 671–676. [Google Scholar] [PubMed]
- World Health Organization. Available online: https://www.who.int/topics/deafness/ru/ (accessed on 2 October 2020).
- Yang, T.; Guo, L.; Wang, L.; Yu, X. Diagnosis, Intervention, and Prevention of Genetic Hearing Loss. Adv. Exp. Med. Biol. 2019, 1130, 73–92. [Google Scholar]
- Online Mendelian Inheritance in Man. Available online: http://www.ncbi.nlm.nih.gov/OMIM (accessed on 31 March 2021).
- Shearer, A.; Hildebrand, M.S.; Smith, R.J.H. Hereditary Hearing Loss and Deafness Overview. GeneReviews® [Internet] Initial Posting: 14 February 1999; Last Update: 27 July 2017. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1434/ (accessed on 2 October 2020).
- Van Camp, G.; Willems, P.J.; Smith, R.J. Nonsyndromic hearing impairment: Unparalleled heterogeneity. Am. J. Hum. Genet. 1997, 60, 758–764. [Google Scholar]
- Morton, N.E. Genetic epidemiology of hearing impairment. Ann. N. Y. Acad. Sci. 1991, 630, 16–31. [Google Scholar] [CrossRef]
- Tekin, M.; Arnos, K.S.; Pandya, A. Advances in hereditary deafness. Lancet 2001, 358, 1082–1090. [Google Scholar] [CrossRef]
- Lee, S.W.; Tomasetto, C.; Paul, D.; Keyomarsi, K.; Sager, R. Transcriptional downregulation of gap-junction proteins blocks junctional communication in human mammary tumor cell lines. J. Cell Biol. 1992, 118, 1213–1221. [Google Scholar] [CrossRef]
- Kenneson, A.; Van Naarden Braun, K.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef]
- Dzhemileva, L.U.; Barashkov, N.A.; Posukh, O.L.; Khusainova, R.I.; Akhmetova, V.L.; Kutuev, I.A.; Gilyazova, I.R.; Tadinova, V.N.; Fedorova, S.A.; Khidiyatova, I.M.; et al. Carrier frequency of GJB2 gene mutations c.35delG, c.235delC and c.167delT among the populations of Eurasia. J. Hum. Genet. 2010, 55, 749–754. [Google Scholar] [CrossRef]
- Zinchenko, R.A.; El’chinova, G.I.; Baryshnikova, N.V.; Polyakov, A.V.; Ginter, E.K. Prevalences of hereditary diseases in different populations of Russia. Russ. J. Genet. 2007, 43, 1038–1045. [Google Scholar] [CrossRef]
- Zinchenko, R.A.; Ginter, E.K.; Kutsev, S.I. Features of the diversity of hereditary diseases in different regions and multiethnic populations of the Russian Federation. Med. Genet. 2020, 19, 13–14. (In Russian) [Google Scholar]
- Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Kadyshev, V.V.; Käsmann-Kellner, B.; Pozdeyeva, N.A.; Katargina, L.A.; Kutsev, S.I.; Zinchenko, R.A. Analysis of Genotype–Phenotype Correlations in Aniridia. J. Med. Genet. 2020. [Google Scholar] [CrossRef]
- Petrova, N.V.; Kashirskaya, N.Y.; Vasilyeva, T.A.; Kondratyeva, E.I.; Zhekaite, E.K.; Voronkova, A.Y.; Sherman, V.D.; Galkina, V.A.; Ginter, E.K.; Kutsev, S.I.; et al. Analysis of CFTR Mutation Spectrum in Ethnic Russian Cystic Fibrosis Patients. Genes 2020, 11, 554. [Google Scholar] [CrossRef] [PubMed]
- Zinchenko, S.P.; Kirillov, A.G.; Abrukova, A.V.; Sorokina, T.V.; Sharonova, E.I.; Khidiyatova, I.M.; Jemileva, L.U.; Shokarev, R.A.; Bliznetz, E.A.; Zinchenko, R.A.; et al. Epidemiology study of hereditary deafness (nonsyndromal and syndromal) Chuvashia Republic. Med. Genet. 2007, 6, 18–28. (In Russian) [Google Scholar]
- Petrina, N.E.; Bliznetz, E.A.; Zinchenko, R.A.; Makaov, A.K.; Petrova, N.A.; Vasilyeva, T.A.; Chudakova, L.V.; Petrin, A.N.; Polyakov, A.V.; Ginter, E.K. The frequency of GJB2 gene mutations in patients with hereditary non-syndromic sensoneural hearing loss in eight populations of Karachay-Cherkess Republic. Med. Genet. 2017, 16, 19–25. (In Russian) [Google Scholar]
- Petrina, N.E.; Bliznetz, E.A.; Zinchenko, R.A.; Makaov, A.Ê.-M.; Petrova, N.A.; Vasilyeva, T.A.; Chudakova, L.V.; Petrin, A.N.; Polyakov, A.V.; Ginter, E.K. Analysis of pedigrees with nonsyndromic sensorineural hearing loss in assortative marriages. Med. Genet. 2018, 17, 47–50. [Google Scholar]
- Shokarev, R.A.; Amelina, S.S.; Kriventsova, N.V.; El’chinova, G.I.; Khlebnikova, O.V.; Bliznetz, E.A.; Tverskaya, S.M.; Polyakov, A.V.; Zinchenko, R.A. Genetic epidemiological and medical genetic study of hereditary deafness in Rostov Oblast. Med. Genet. 2005, 4, 556–565. (In Russian) [Google Scholar]
- Sharonova, E.I.; Osetrova, A.A.; Zinchenko, R.A. Hereditary hearing disorders in Kirov Oblast. Yakut Med. J. 2009, 2, 28–31. (In Russian) [Google Scholar]
- Zinchenko, R.A.; Osetrova, A.A.; Sharonova, E.I. Hereditary Deafness in Kirov Oblast: Estimation of the Incidence Rate and DNA Diagnosis in Children. Russ. J. Genet. 2012, 48, 542–550. [Google Scholar] [CrossRef]
- Zinchenko, R.A.; Makaov, A.K.; Kadyshev, V.V.; Galkina, V.A.; Dadali, E.L.; Mikhailova, L.K.; Shurygina, M.F.; Marakhonov, A.V.; Petrova, N.V.; Petrona, N.E.; et al. Diversity and prevalence of hereditary diseases among Nogais of the Karachay-Cherkess Republic. Russ. J. Genet. 2018, 54, 858–865. [Google Scholar] [CrossRef]
- Petrina, N.E.; Marakhonov, A.V.; Balinova, N.V.; Mironovich, O.L.; Polyakov, A.V.; Zinchenko, R.A. Description of a rare case of hereditary hearing loss with X-linked recessive type of inheritance associated with the POU3F4 gene in the Nogai family. Vestn. Otorhinolaryngol. 2020, 85, 65–69. [Google Scholar] [CrossRef]
- Lozier, E.R.; Konovalov, F.A.; Kanivets, I.V.; Pyankov, D.V.; Koshkin, P.A.; Baleva, L.S.; Sipyagina, A.E.; Yakusheva, E.N.; Kuchina, A.E.; Korostelev, S.A. De novo nonsense mutation in WHSC1 (NSD2) in patient with intellectual disability and dysmorphic features. J. Hum. Genet. 2018. [Google Scholar] [CrossRef]
- Zhivotovsky, L.A. Population-Based Biometrics; Nauka: Moscow, Russia, 1991. (In Russian) [Google Scholar]
- Clopper, C.J.; Pearson, S. The use of confidence or fiducial limits illustrated in the canse of the binomial. Biometrika 1934, 26, 404–413. [Google Scholar] [CrossRef]
- The Genome Aggregation Database. Available online: http://gnomad.broadinstitute.org (accessed on 31 March 2021).
- Charizopoulou, N.; Lelli, A.; Schraders, M.; Ray, K.; Hildebrand, M.S.; Ramesh, A.; Srisailapathy, C.R.; Oostrik, J.; Admiraal, R.J.; Neely, H.R.; et al. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat. Commun. 2011, 15, 201. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Gul, K.; Morell, R.J.; Lee, K.; Ahmed, Z.M.; Riazuddin, S.; Ali, R.A.; Shahzad, M.; Jaleel, A.U.; Andrade, P.B.; et al. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p. Hum. Genet. 2011, 130, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Ramzan, K.; Al-Owain, M.; Allam, R.; Berhan, A.; Abuharb, G.; Taibah, K.; Imtiaz, F. Homozygosity mapping identifies a novel GIPC3 mutation causing congenital nonsyndromic hearing loss in a Saudi family. Gene 2013, 521, 195–199. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Horta, O.; Duman, D.; Foster, J., 2nd; Sırmacı, A.; Gonzalez, M.; Mahdieh, N.; Fotouhi, N.; Bonyadi, M.; Cengiz, F.B.; Menendez, I.; et al. Whole-exome sequencing efficiently detects rare mutations in autosomal recessive nonsyndromic hearing loss. PLoS ONE 2012, 7, e50628. [Google Scholar]
- Sirmaci, A.; Edwards, Y.J.; Akay, H.; Tekin, M. Challenges in whole exome sequencing: An example from hereditary deafness. PLoS ONE 2012, 7, e32000. [Google Scholar] [CrossRef]
- Asgharzade, S.; Tabatabaiefar, M.A.; Mohammadi-Asl, J.; Chaleshtori, M.H. A novel missense mutation in GIPC3 causes sensorineural hearing loss in an Iranian family revealed by targeted next-generation sequencing. Int. J. Pediatr. Otorhinolaryngol. 2018, 108, 8–11. [Google Scholar] [CrossRef]
- Ahmad, W.; Zhang, X. Sequence variants in genes causing nonsyndromic hearing loss in a Pakistani cohort. Mol. Genet. Genom. Med. 2019, 7, e917. [Google Scholar]
- Ammar-Khodja, F.; Bonnet, C.; Dahmani, M.; Ouhab, S.; Lefèvre, G.M.; Ibrahim, H.; Hardelin, J.P.; Weil, D.; Louha, M.; Petit, C. Diversity of the causal genes in hearing impaired Algerian individuals identified by whole exome sequencing. Mol. Genet. Genom. Med. 2015, 3, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Bitarafan, F.; Seyedena, S.Y.; Mahmoudi, M.; Garshasbi, M. Identification of novel variants in Iranian consanguineous pedigrees with nonsyndromic hearing loss by next-generation sequencing. J. Clin. Lab. Anal. 2020, 34, e23544. [Google Scholar] [CrossRef]
- Kannan-Sundhari, A.; Yan, D.; Saeidi, K.; Sahebalzamani, A.; Blanton, S.H.; Liu, X.Z. Screening Consanguineous Families for Hearing Loss Using the MiamiOtoGenes Panel. Genet. Test Mol. Biomark. 2020, 24, 674–680. [Google Scholar] [CrossRef]
- Zhou, Y.; Tariq, M.; He, S.; Abdullah, U.; Zhang, J.; Baig, S.M. Whole exome sequencing identified mutations causing hearing loss in five consanguineous Pakistani families. BMC Med. Genet. 2020, 21, 151. [Google Scholar] [CrossRef]
- Alekseev, V. The Geography of Human Races, 1st ed.; Nauka: Moscow, Russia, 1974. [Google Scholar]
- Kuzeev, R. The Peoples of the Volga and Ural Regions, 1st ed.; Nauka: Moscow, Russia, 1985. [Google Scholar]
- Bliznetz, E.A.; Tverskaya, S.M.; Zinchenko, R.A.; Abrukova, A.V.; Savaskina, E.N.; Nikulin, M.V.; Kirillov, A.G.; Ginter, E.K.; Polyakov, A.V. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: The unique splice site mutation in TCIRG1 gene spread by the founder effect. Eur. J. Hum. Genet. 2009, 17, 664–672. [Google Scholar] [CrossRef]
- Vasserman, N.N.; Zinchenko, R.A.; Polyakov, A.V. The age of Arg200Trp mutation in VHL gene, the cause of autosomal recessive erythrocytosis in Chuvashia. Med. Genet. 2007, 6, 31–36. (In Russian) [Google Scholar]
- Kazantseva, A.; Goltsov, A.; Zinchenko, R.; Grigorenko, A.P.; Abrukova, A.V.; Moliaka, Y.K.; Kirillov, A.G.; Guo, Z.; Lyle, S.; Ginter, E.K.; et al. Human Hair Growth Deficiency Is Linked to a Genetic Defect in the Phospholipase Gene LIPH. Science 2006, 314, 982–985. [Google Scholar] [CrossRef]
- Zernov, N.V.; Skoblov, M.Y.; Marakhonov, A.V.; Shimomura, Y.; Vasilyeva, T.A.; Konovalov, F.A.; Abrukova, A.V.; Zinchenko, R.A. Autosomal Recessive Hypotrichosis with Woolly Hair Caused by a Mutation in the Keratin 25 Gene Expressed in Hair Follicles. J. Investig. Dermatol. 2016, 136, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, A.A.; Abrukova, A.V.; Savaskina, E.N.; Polyakov, A.V. Mutation p.E92K is the Primary Cause of Cystic Fibrosis in Chuvashes. Russ. J. Genet. 2012, 48, 731–737. [Google Scholar] [CrossRef]




| Family # | Patient’s Age | Age of Onset | Age of Manifestation | Degree of Hearing Loss | Genotype for GIPC3 Coding Region |
|---|---|---|---|---|---|
| 01 | 24 | Early childhood | noticed in 2 years | Bilateral; severe | NM_133261.3(GIPC3):c.[245A>G];[245A>G] (p.[Asn82Ser];[Asn82Ser]) |
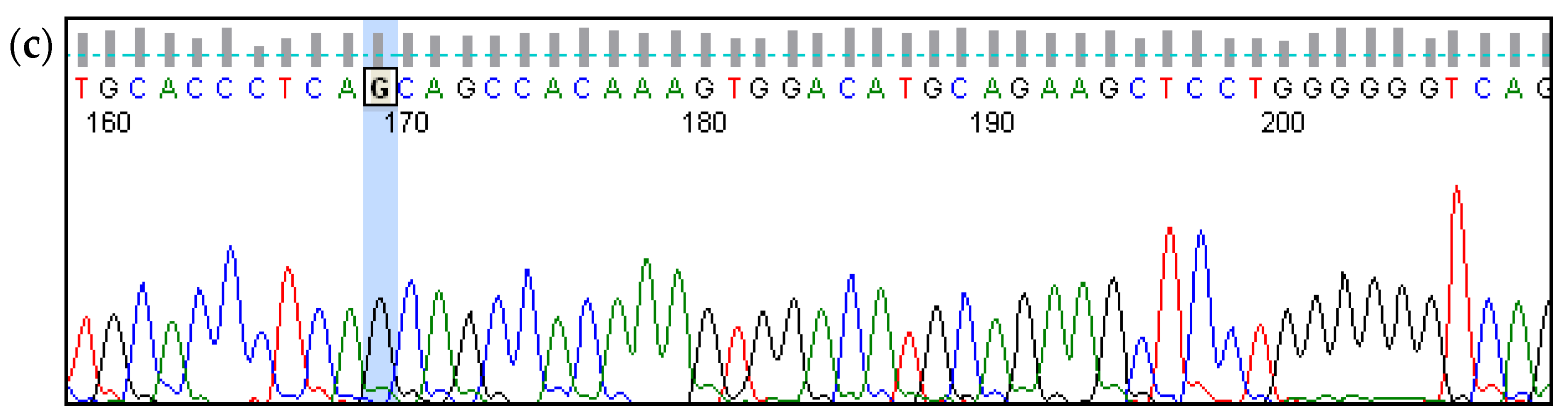
| 02 | 14 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 03 | 8 | Congenital | from the first months of life | Bilateral; severe. | NM_133261.3(GIPC3):c.[=];[=] |
| 04 | 14 | Congenital | at 5 months | Bilateral; complete | NM_133261.3(GIPC3):c.[245A>G];[=] (p.[Asn82Ser];[=]) |
| 05 | 18 | Congenital | noticed in 2 years | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 06 | 33 | Congenital | noticed in 2 years | Bilateral; severe | NM_133261.3(GIPC3):c.[=];[=] |
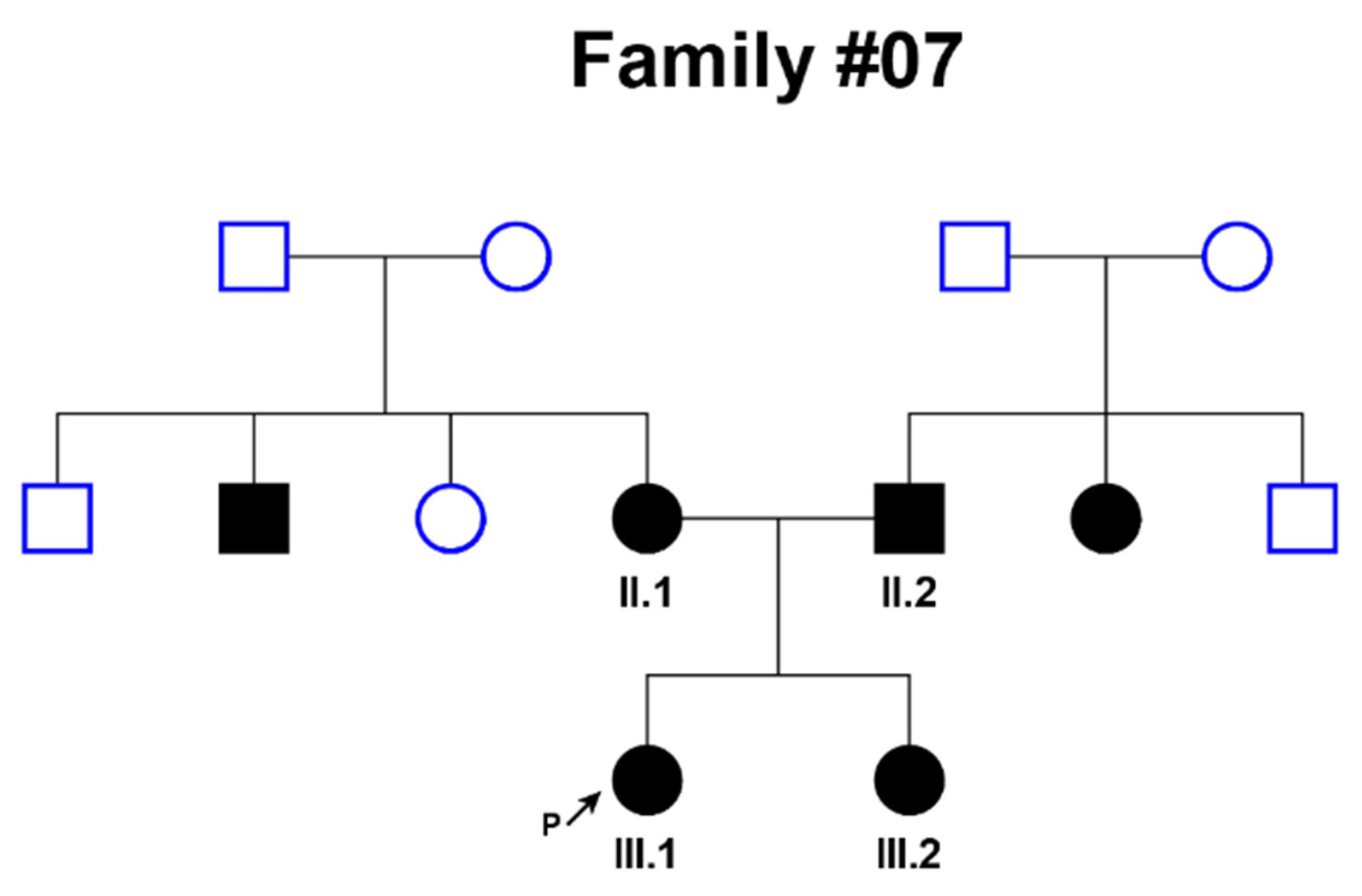
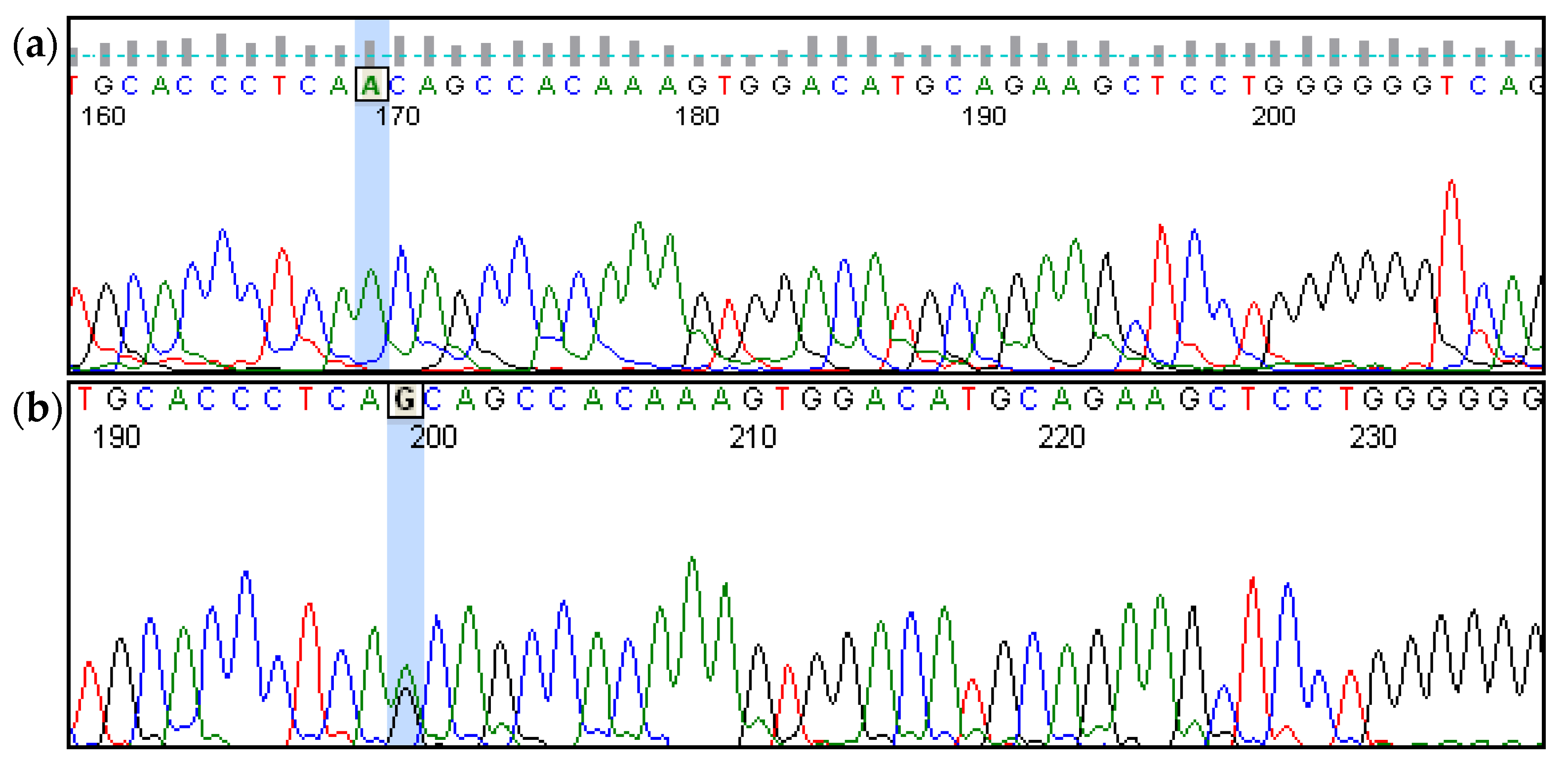
| 07 | 1 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[245A>G];[245A>G] (p.[Asn82Ser];[Asn82Ser]) |
| 07 | 28 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[245A>G];[245A>G] (p.[Asn82Ser];[Asn82Ser]) |
| 07 | 32 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[245A>G];[245A>G] (p.[Asn82Ser];[Asn82Ser]) |
| 07 | 4 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[245A>G];[245A>G] (p.[Asn82Ser];[Asn82Ser]) |
| 08 | 12 | Congenital | noticed in 3 years | OD-moderate, OS severe | NM_133261.3(GIPC3):c.[=];[=] |
| 09 | 10 | Congenital | noticed in 1 year | OD-moderate, OS severe | NM_133261.3(GIPC3):c.[=];[=] |
| 10 | 18 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 11 | 14 | Congenital | from the first months of life | OD-severe, OS profound | NM_133261.3(GIPC3):c.[=];[=] |
| 12 | 6 | Congenital | noticed in 3 years | Bilateral; profound. | NM_133261.3(GIPC3):c.[=];[=] |
| 13 | 10 | Congenital | from the first months of life | OD-severe, OS profound | NM_133261.3(GIPC3):c.[=];[=] |
| 14 | 15 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 15 | 16 | Congenital | noticed in 1.5 years | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 16 | 19 | Congenital | from the first months of life | Bilateral; complete | NM_133261.3(GIPC3):c.[=];[=] |
| 17 | 14 | Childhood | 5–6 years | Unilateral; OD moderate | NM_133261.3(GIPC3):c.[=];[=] |
| 17 | 42 | Childhood | 13–14 years | Unilateral; OS moderate to severe | NM_133261.3(GIPC3):c.[=];[=] |
| 18 | 13 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 18 | 38 | Congenital | noticed in 1.5 years | Bilateral; profound | NM_133261.3(GIPC3):c.[=];[=] |
| 19 | 36 | Congenital | noticed by 6–7 months | OD complete, OS profound | NM_133261.3(GIPC3):c.[=];[=] |
| 20 | 22 | Congenital | from the first months of life | Bilateral; profound | NM_133261.3(GIPC3):c.[245A>G];[245A>G] (p.[Asn82Ser];[Asn82Ser]) |
| 21 | 22 | Congenital | noticed by 12–13 months | OD moderate, OS severe | NM_133261.3(GIPC3):c.[=];[=] |
| Gene | Primer Name and Sequence in 5′—3′ Direction |
|---|---|
| GJB2 | F1 TCATGGGGGCTCAAAGGAAC |
| R1 AAGGACGTGTGTTGGTCCAG | |
| F2 GTTCTGTCCTAGCTAGTGATT | |
| R2 GGTTGCCTCATCCCTCTCAT | |
| GJB3 | F1 CGTTGTGAGTATTGAACAAGTCAGAACTCAG |
| R1 GTTGATCCCTTCCTGGTTA | |
| F2 CTCTGCTACCTCATCTGCCA | |
| R2 GTTGATCCCTTCCTGGTTGA | |
| GJB6 | F1 CTTTCAGGGTGGGCATTCCT |
| R1 AGCACAACTCTGCCACGTTA | |
| F2 CTTCGTCTGCAACACACTGC | |
| R2 GCAATGCTCCTTTGTCAAGCA | |
| GIPC3-ex1 | F1 CTTATTTGTGGTCCCTGTTCTTC |
| R1 AGTCCTAAGACCTGCCCATCT | |
| GIPC3-ex2–4 | F2 CTCTCTCTGTTCTGGGGGTCC |
| R2 ACCTACGAGTTTCTGATACCCTG | |
| GIPC3-ex5–6 | F3 GGCATGGAACTGGGATGTTA |
| R3 GCACATAGCTTGGCCTCAGAT | |
| GIPC3-c.245A>G | GIPC3-Fmut TCTCCACCTGCTGGAAGTCT |
| GIPC3-Rmut CCTCGATCCGGTTGATGAT |
| Single Nucleotide Polymorphism (SNP) | rs112835547 | rs34722692 | rs8100350 | rs8113232 | rs4806942 | rs10406702 | rs10426399 | rs28532669 | rs78077103 | rs78077103 | rs17348907 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Possible genotypes (ref/alt) | C/T | C/T | G/A | A/G | A/G | T/C | C/T | G/A | G/T | G/A | C/T |
| Haplotype linked to the variant | T | C | G | G | G | T | C | G | G | G | T |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrova, N.V.; Marakhonov, A.V.; Balinova, N.V.; Abrukova, A.V.; Konovalov, F.A.; Kutsev, S.I.; Zinchenko, R.A. Genetic Variant c.245A>G (p.Asn82Ser) in GIPC3 Gene Is a Frequent Cause of Hereditary Nonsyndromic Sensorineural Hearing Loss in Chuvash Population. Genes 2021, 12, 820. https://doi.org/10.3390/genes12060820
Petrova NV, Marakhonov AV, Balinova NV, Abrukova AV, Konovalov FA, Kutsev SI, Zinchenko RA. Genetic Variant c.245A>G (p.Asn82Ser) in GIPC3 Gene Is a Frequent Cause of Hereditary Nonsyndromic Sensorineural Hearing Loss in Chuvash Population. Genes. 2021; 12(6):820. https://doi.org/10.3390/genes12060820
Chicago/Turabian StylePetrova, Nika V., Andrey V. Marakhonov, Natalia V. Balinova, Anna V. Abrukova, Fedor A. Konovalov, Sergey I. Kutsev, and Rena A. Zinchenko. 2021. "Genetic Variant c.245A>G (p.Asn82Ser) in GIPC3 Gene Is a Frequent Cause of Hereditary Nonsyndromic Sensorineural Hearing Loss in Chuvash Population" Genes 12, no. 6: 820. https://doi.org/10.3390/genes12060820
APA StylePetrova, N. V., Marakhonov, A. V., Balinova, N. V., Abrukova, A. V., Konovalov, F. A., Kutsev, S. I., & Zinchenko, R. A. (2021). Genetic Variant c.245A>G (p.Asn82Ser) in GIPC3 Gene Is a Frequent Cause of Hereditary Nonsyndromic Sensorineural Hearing Loss in Chuvash Population. Genes, 12(6), 820. https://doi.org/10.3390/genes12060820