Abstract
Kallmann syndrome (KS) is a combination of isolated hypogonadotropic hypogonadism (IHH) with olfactory dysfunction, representing a heterogeneous disorder with a broad phenotypic spectrum. The genetic background of KS has not yet been fully established. This study was conducted on 46 Polish KS subjects (41 males, 5 females; average age: 29 years old). The studied KS patients were screened for defects in a 38-gene panel with next-generation sequencing (NGS) technology. The analysis revealed 27 pathogenic and likely pathogenic (P/LP) variants, and 21 variants of uncertain significance (VUS). The P/LP variants were detected in 20 patients (43.5%). The prevalence of oligogenic P/LP defects in selected genes among KS patients was 26% (12/46), whereas the co-occurrence of other variants was detected in 43% (20 probands). The examined KS patients showed substantial genotypic and phenotypic variability. A marked difference in non-reproductive phenotypes, involving defects in genes responsible for GnRH neuron development/migration and genes contributing to pituitary development and signaling, was observed. A comprehensive gene panel for IHH testing enabled the detection of clinically relevant variants in the majority of KS patients, which makes targeted NGS an effective molecular tool. The significance of oligogenicity and the high incidence of alterations in selected genes should be further elucidated.
1. Introduction
Kallmann syndrome (KS) is a rare hormonal disorder of sexual maturation and fertility. Smell disturbances (hyposmia or anosmia) are an indispensable part of KS. Patients with KS constitute approximately 60% of subjects with isolated hypogonadotropic hypogonadism (IHH) [1]. Gonadotropin-releasing hormone (GnRH) neuron development and migration disturbances or dysfunction represent the core background of the disease. The origin of GnRH neurons is a nasal placode—an embryonal region that invaginates to establish the olfactory epithelium and the vomeronasal organ, giving rise to the formation of olfactory sensory neurons and olfactory ensheathing cells [2]. During embryogenesis, GnRH neurons from the vomeronasal organ travel along the axons surrounded by olfactory ensheathing cells to the forebrain—a final destination for governing hormone signaling through the hypothalamic–pituitary–gonadal (HPG) axis and contributing to proper sexual advancement [3]. The common origin of GnRH and olfactory neurons, and disturbances in the development of olfactory placodes, deliver reasonable explanation for the associated occurrence of impaired sense of smell (anosmia, hyposmia) in KS patients comparing to other IHH forms. KS might be also accompanied by diverse congenital defects, including midline cranial anomalies, dental agenesis, renal defects, and limb malformations [4,5,6]; its most complex form is known as CHARGE syndrome—a congenital disorder associated with coloboma, heart malformation, choanal atresia, retardation of growth and/or development, genital anomalies, and ear anomalies/deafness. Because of the overlapping clinical manifestation, IHH and olfactory defects are today considered to be part of CHARGE syndrome [7,8]. This phenotype suggests the involvement of broader complex embryonic disturbances than those related to the HPG axis, and various cellular processes [9]. So far, more than 50 genes involved in the development of the hypothalamus and pituitary gland, formation and migration of GnRH neurons, or the regulation of GnRH and gonadotropin secretion have been described. Due to the contribution of genes located on the X chromosome, hypogonadotropic hypogonadism is five times more common in males than in females. About 1 in 30,000 men and 1 in 125,000 women are affected by KS [10,11,12]. The inheritance patterns for KS encompass X-linked recessive, autosomal dominant, and autosomal recessive transmission. Digenic and oligogenic defects are increasingly reported [13,14], constituting a major explanation for the clinical variability of the disorder. Moreover, the genetic background for barely half of IHH cases is established, and new genetic targets are still being sought for IHH patients [14,15,16]. KS/IHH is a genetically and phenotypically heterogeneous disease whose incidence, as well as the phenotypic and genetic spectrum of KS/IHH for the Polish population, is limited [17,18,19,20,21,22,23].
Objective
The current study aimed to investigate the genetic basis of KS in adult Polish patients. The study also sought to report the ratios of pathogenic or likely pathogenic (P/LP) alterations, variants of uncertain significance (VUS), and benign or likely benign (B/LB) alterations, according to the American College of Medical Genetics and Genomics (ACMG)’s classification. The objective of the study was to establish genotype–phenotype correlations, as well as to identify variant recurrence among KS patients in Poland.
2. Materials and Methods
2.1. Patients
A group of 46 well-characterized patients (41 males, 5 females; average age: 29; min–max: 18–57 years) with KS were included in the study and subjected to genomic screening. Phenotypic information was based on clinical interview and physical examination, including anthropometric measurements. The history of unilateral or bilateral cryptorchidism was collected. The patients were subjected to an andrological assessment (physical examination and ultrasound examination of the testicles) or a gynecological examination. In each case, an abdominal ultrasound examination was conducted in order to detect additional congenital defects. The reversibility of KS in male subjects was assessed as the capability to achieve a normal adult testosterone serum concentration after six months of hormone replacement therapy withdrawal [24]. Pubertal status was specified as lack of puberty, incomplete puberty, or complete puberty [25].
To assess pituitary function, the measurements of basic and, if necessary, stimulated concentrations of pituitary hormones—luteinizing and follicle-stimulating hormones (LH, FSH), growth hormone (GH), adrenocorticotropic hormone (ACTH), thyrotropin (TSH)s and prolactin (PRL)—were carried out. Measurements of testosterone, estradiol, morning cortisol, dehydroepiandrosterone sulfate (DHEA-S), insulin-like growth factor 1 (IGF-1), free triiodothyronine (fT3), and free thyroxin (fT4) in fasting blood serum were also taken. Serum cortisol level was also measured at 6:00 p.m. Hormonal measurements were performed with a Cobas 6000 (Roche Diagnostics, Basle, Switzerland) using dedicated electrochemiluminescence sandwich immunoassay (ECLIA) kits provided by the manufacturer. The following calculation determined the free testosterone index (FTI): (FTI) = 100 × (total testosterone/SHBG). A test with 100 μg of intravenous gonadoliberin (LHRH, Ferring) was conducted in each patient. Hypogonadotropic hypogonadism was diagnosed in all patients according to the European Consensus Statement (2015), and was based on the presence of low serum levels of testosterone in male individuals (3.5 nmol/L) or low estradiol levels (<20 pg/mL) in females, associated with serum levels of gonadotropins of less than 5 international units/L, without reaction to GnRH [25].
No other associated pituitary endocrinopathy was identified in the studied subjects. The body weight and height of the patients were measured using a height-measuring stand with a weight and height scale machine to obtain the anthropometric data. BMI was calculated according to the Quetelet formula: BMI (kg/m2) = weight (kg)/height (m2).
All patients underwent ENT assessment and psychophysical testing of the olfactory function. Olfactometry was performed according to Elsberg and Levy’s method, modified by Pruszewicz [26,27]. Elsberg and Levy’s method, enabling detection and recognition thresholds for the four odorants, is the most widely used and available smell-testing method in Poland. All patients were checked for presentation of any features of the CHARGE spectrum.
2.2. Genetic Studies
Molecular studies were conducted using next-generation sequencing (NGS) technology with the Ion Torrent Personal Genome Machine system (Ion PGM ™, Thermo Fisher Scientific, Waltham, MA, USA). The targeted panel of 38 genes associated with IHH and combined pituitary hormone deficiency (CPHD) encompassed: ADAM7, ANOS1, BMP2, BMP4, CHD7, FGF8, FGF17, FGFR1, GLI2, GNRH1, GNRHR, HESX1, HS6ST1, IGSF10, KISS1, KISS1R, LEP, LEPR, LHB, LHX3, LHX4, LRRIQ3, NSMF, NR0B1, OTX1, OTX2, PCSK1, PITX1, PITX2, PROK2, PROKR2, PROP1, POU1F1, SEMA3A, SOX3, TAC3, TACR3, and WDR11.
The sequences were mapped to the human genome (GRCh37/hg19) using Torrent Suite ™ software (version 4.0.2, Thermo Fisher Scientific, Waltham, MA, USA), and the variant positions were then translated to (GRCh38/hg38) when needed. Each detected variant was assigned according to HGVS nomenclature with the use of canonical transcripts (according to Ensembl). The analysis of the obtained sequences was made using the following algorithms: PhenIX (http://compbio.charite.de/PhenIX/), Mutation Taster 2 (http://www.mutationtaster.org/), and Variant Effect Predictor (VEP) in Ensembl (19), assuming prediction from FATHMM, CAD, PolyPhen2, SIFT, and MaxEntScan. For pathogenicity evaluation, the clinical databases HGMD (http://www.hgmd.cf.ac.uk) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) were also checked. The GnomAD database (https://gnomad.broadinstitute.org/) was explored to establish the reported presence and frequency of variants. The minor allele frequency (MAF) of < 1% in GnomAD was assumed as the primary criterion for pathogenicity examination. Alterations were classified according to updated guidelines reported by the ACMG—pathogenic and/likely pathogenic (P/LP), variant of unknown clinical significance (VUS), and benign and/likely benign (B/LB)—assuming impact on protein functionality, evolutionary conservativeness, and frequency. Varsome [28] and InterVar [29] were used for correctness of position estimation and HGVS nomenclature. The detected mutations were confirmed using conventional capillary sequencing. Selected mutations were also subjected to in silico analysis in order to evaluate the effect of the change on protein architecture, considering its functionality and potential involvement in the manifestation of an abnormal phenotype using PHYRE2 (http://www.sbg.bio.ic.ac.uk) and Chimera v1.7 [30].
3. Results
3.1. Characteristics of the Group
The analyzed KS group had a high male predominance (M:F ratio of 8:1). Four familial cases of KS were included in the study. A severe reproductive phenotype was observed in the majority of patients. The prevalence of a total lack of puberty reached 78%. Incomplete puberty was reported in 9% of subjects. In contrast, complete puberty was seen in 13% of patients. Hyposmia was present in 28% (13/46) of patients, and 58% (27/46) of patients were anosmic, according to the psychophysical smell test. The 36% (15/41) of male patients had diagnosed cryptorchidism—unilateral in six cases and bilateral in nine patients. The prevalence of the additional congenital defects, apart from dysosmia, was 45%. The most common abnormalities included renal defects (renal agenesis, horseshoe kidney, double pyelum; five cases), limb malformations (syndactyly, clinodactyly, brachydactyly; five cases), and mirror movements (three cases). The incidence of reversible IHH forms was noted for 6% of cases (three cases). Detailed clinical data are presented in Table 1 and Table 2.

Table 1.
KS patients with genetic defects in primary GnRH neuron development/migration.
Table 2.
KS patients with genetic defects in hypothalamic/pituitary development and signaling.
3.2. Genetic Results
Genetic defects were classified as those related to GnRH neuron development and migration (Table 1), and those related to pituitary development and signaling (Table 2). Defects in 18 genes were detected for 46 KS patients. The 50 identified variants were re-graded as clinically relevant, of which 11 were classified as pathogenic (P), 12 as likely pathogenic (LP), 16 as VUS, and 11 as benign or likely benign (B/LB) (Figure 1A). Heterozygous P/LP alterations were found in the CHD7 (n=7; 15.2% of all KS subjects, all novel and not reported); FGFR1 (n = 6; 13%, 5 novel), GNRHR (n = 4; 9%, 2 novel), GNRH1 (n = 1; 2%), WDR11 (n = 1; 2%), PROKR2 (n = 1; 2%), GLI2 (n = 1; 2%), and LHX4 (n = 1; 2%) genes. Hemizygous P/LP variants were detected in the ANOS1 (n = 1, familial case (two patients); 2%) and SOX3 (n = 1; 2%) genes (Table 1 and Table 2). For six IHH patients (two with hyposmia and four with anosmia), no causative defects were identified at all. Monogenic variants with clinically significant P/LP alteration were seen in eight KS patients and in another seven subjects presenting oligogenicity (Figure 1B). In total, 15 novel pathogenic variants, not previously reported for KS, were detected in 19 patients affecting 7 genes (ANOS1, CHD7, FGFR1, GLI2, GNRHR, SOX3, and WDR11) (Figure 1C).
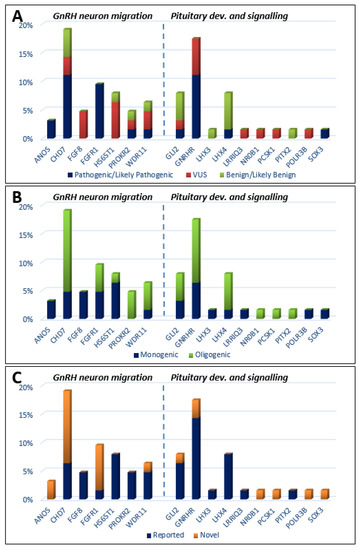
Figure 1.
Histograms showing the prevalence of mutations among KS patients, which are classified by three metrics: (A) mutation severity (ACMG criteria: pathogenic/likely pathogenic, VUS variant of unknown significance, benign/likely benign); (B) oligogenicity; (C) novelty.
Concerning variant recurrence, the heterozygous mutation in FGFR1 p.R285W (likely pathogenic; CM063987 HGMD), coexisting with another heterozygous variant CHD7 p.N1030H (pathogenic; novel), was recurring in two unrelated patients, both with reversal of hypogonadism. A heterozygous variant in LHX4 p.D128= (benign; MAF 0.006, rs141139762) was detected in three cases (four patients, including two affected brothers). In summary, the applied approach identified 18 P/LP variants in genes responsible for the development and migration of GnRH neurons (Table 1) and 9 P/LP variants in genes contributing to pituitary formation and pituitary/hypothalamic signaling (Table 2). P/LP variants in pituitary genes were identified for two of the examined KS patients: an oligogenic defect in LHX4 (p.G305W)/CHD7 (p.K850Q), and proband 34 with a p.R155Afs*26 defect in SOX3 (Table 1 and Table 2). Detailed information referring to identified variants, comprising their precise genomic coordinates and functional prediction, is included in the Table S1. In order to further explore the involvement of HS6ST1 (p.D87E, p.R249S) and PROKR2 (p. R85H, p.R268C) variants, in silico modelling was performed. Except for HS6ST1p.R249S (low confidence of prediction), changes in protein architecture were noted for all mentioned variants (Figure 2).
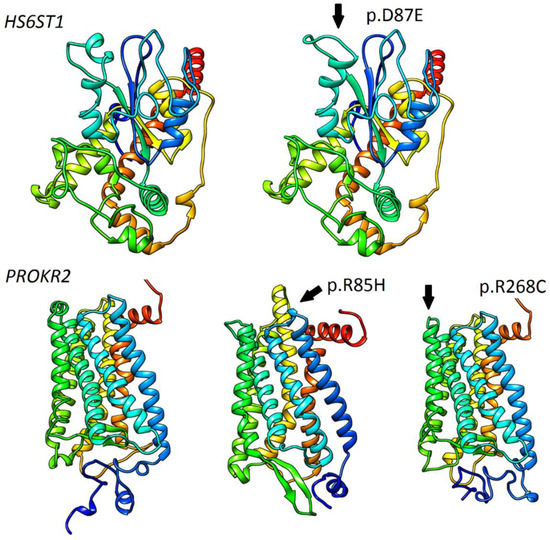
Figure 2.
Modelling and prediction of conformational changes of selected HS6ST1 and PROKR2 variants. Predicted conformational changes surrounding variant positions are highlighted and indicated with arrows. Ribbon models of reference HS6ST1 and PROKR2 are shown on the left side.
3.3. Genotype–Phenotype Correlations
Associated non-reproductive malformations were much more common in patients with defects in the genes governing GnRH neuron development and migration than in patients with defects associated with pituitary and signaling cascades (Table 1 and Table 2). Limb malformations were recorded only in the P/LP-variants-positive group, with defects in CHD7, FGFR1, HS6ST1, and LHX4. The majority of patients with digital bone abnormalities presented heterozygous mutation in CHD7 (80% of cases). Similarly, renal abnormalities were detected only in the P-variant-positive group (six patients with mutations in ANOS1, CHD7, FGFR1, and GNRHR). Synkinesis was seen in three cases with mutated ANOS1 (family case), FGF8, and digenic defects in GNRHR and WDR11. Dental agenesis was detected in the male patients with digenic defects in CHD7 and LHX4 (proband 8). Additional CHARGE features were present in two analyzed KS patients with P/LP variants in CHD7: proband 13 (choanal atresia), and proband 21 (ear anomaly).
3.4. Male Reversibility
In a long-term clinical follow-up, three unrelated subjects with digenic defects (proband 9: CHD7 p.N1030H and FGFR1 p.R285W; proband 10: CHD7 p.N1030H and FGFR1 p.R285W; and proband 25: GNRHR p.R139H, GNRHR p.N10_Q11delinsKK, and CHD7 p.K683_T684insAK) displayed a reversal of hypogonadism. Two of them had hyposmia. Only one of them had an additional congenital deformity (sandal gap) and a KS family history.
3.5. Females
In the current cohort, P/LP variants were found in two out of five female patients, affecting the GNRH1 and GNRHR genes. The two anosmic KS females with a history of primary amenorrhea harbored P/LP variants (in the first case in the GNRH1 gene (with no other accompanying congenital defect), and in the second case in the GNRHR gene), presenting incomplete rotation of the right kidney. (Table 2)
4. Discussion
4.1. Effectiveness of Panel-Based NGS
The study was conducted on 46 adult patients with Kallmann syndrome. The panel-based NGS approach used in this study appears to be an efficient and cost-effective method. A panel of 38 genes enabled the detection of causative variants (P/LP) in 43.5% of KS patients. The frequency of new P/LP variants was high: 65% of all detected P/LP variants, and in 37% of patients (17/46). VUS alterations were detected in 37% of studied subjects. The failure to find P/LP variants In 56% of enrolled subjects could explain mutations outside the coding regions or mutations in other IHH candidate genes [16].
The overall effectiveness of the application of NGS with a panel of 38 IHH/CPHD genes in the current study was similar to or even higher than the efficacy of the panel-based NGS approach in previous studies. Targeted NGS was also applied by Zhou et al. in 148 Chinese male IHH patients. Zhou et al. used a panel of 83 genes (31 known IHH genes and 52 candidate genes) and identified P/LP variants in 14.9% of patients, in 16 causal genes and 36 candidate genes. The results of targeted NGS of 261 candidate genes in 48 subjects with KS/nIHH with no mutations in any known KS/nIHH genes were described by Quyanor et al., who detected two new LP variants in FGFR1, and proposed 18 new candidate genes in KS/nIHH [31]. Butz et al. employed a panel of 41 genes to analyze the genetic backgrounds of 38 patients with hypogonadotropic hypogonadism, and found genetic defects in 8 patients (21%) [32]. Gach et al. identified known or novel potentially deleterious variants in 23 out of 47 (49%) unrelated IHH patients (31 men, 16 women; 26 nIHH and 23 KS) tested with NGS using a 51-gene panel. In the next step of the Gach et al. study, a WES analysis and molecular diagnosis were completed in 19/47 CHH cases. Monogenic mutations were found in 13 subjects in ANOS1, FGFR1, GNRHR, CHD7, SOX10, and PROKR2, whereas in 6 patients oligogenic variants were identified in SPRY/SEMA3A, CCDC141/POLR3B, SRA1/SEMA7A, CHD7/SEMA71, NSMF/SPRY4/PROKR2, and TACR3/LHB [22]. Cangiano et al. examined 160 males with classic or milder ao-IHH—both nIHH and KS—using NGS and a panel of 28 genes. In that study, rare gene variants were detected in 55% of patients (although it was 32% considering only the 13 first-discovered IHH genes (ANOS1, FGFR1, PROKR2, PROK2, GNRHR, GNRH1, GNRH2, KISS1, KISS1R, TAC3, TACR3, HS6ST1, FGF8)) [33]. The effectiveness of the panel used in the current study was so high due to the careful selection of only patients with KS, as well as the designed NSG gene panel, which covered the most commonly mutated genes.
4.2. Defects in Genes Responsible for GnRH Neuron Migration and Development
Previously reported and new genetic factors involved in the etiopathogenesis of KS have been identified in the studied patients. P/LP variants in genes involved in neuron migration (ANOS1, CHD7, FGFR1, and WDR11) were found predominantly in the current study. The most common P/LP variants in Polish probands were found in the CHD7 and FGFR1 genes. Furthermore, the most severe reproductive phenotype was seen in patients with alterations in those genes, particularly when accompanying variants in other IHH genes were present. The CHD7- and FGFR1-positive patient group was significantly enriched by additional non-reproductive congenital malformations. The 15% prevalence of P/LP variants of CHD7 in Polish KS patients is higher than the reported incidence of 4–10% in other IHH studies [22,34,35]. In addition, the incidence of mutations in FGFR1 appears to be higher than in previous reports—13% vs. 8.5% [22]. The rate of mutations in GNRHR, GNRH1, and PROKR2 was similar to that reported in the literature. In contrast, mutations in WDR11, HS6ST1, and FGF8 were recognized in the current study slightly more often than in previous literature [34,36], although one must consider the number of KS patients analyzed in the current study.
Patients enrolled in the current study were characterized by notable genetic and phenotypic variability. Even the presence of the same P/LP variant appears to result in variable expressivity (e.g., patients 1 and 2 (family case), patients 9 and 10 (unrelated subjects)). The genetic heterogeneity of gene defects in CHD7, FGFR1, and GNRHR was observed in the current study. Genotypic and phenotypic diversity of KS is broad, and varies among different populations, presenting a significant diagnostic challenge. The genetic heterogeneity of KS, especially within respective pedigrees, has been noted by multiple authors [16,37,38].
No recurrent founder mutation originating from Poland was found. The p.R285W defect in FGFR1 was co-occurring with p.N1030H in the CHD7 gene as a digenic case for two sporadic, unrelated patients. It is worth noting that 15 new P/LP variants were detected in known IHH genes, which emphasizes the importance of the comprehensive elucidation of known genes in various populations. An interesting case of co-occurrence of two variants in HS6ST1 (p.D87E, p.R249S) was detected. Because variants were found in two unrelated sporadic patients, we think that this was more likely a monoallelic case (cis form) than compound heterozygosity (trans), but the DNA of the patients’ parents was unavailable, and so we could not check it. Despite the phenotypical manifestation, the lack of other relevant abnormal variants and protein modeling data for p.D87E seems to suggest clinical relevancy. Causativeness for two oligogenic variants in PROKR2 (p.R85H, p.R268C) seems to confirm previous clinical and functional reports [39,40](Figure 2).
4.3. Defects in Genes Responsible for Pituitary and Hypothalamic Development and Signaling
A shared genetic background for CPHD, holoprosencephaly, and IHH/KS was proposed by Raivo et al. and Vaaralahti et al. [41,42]. Rare variants of genes implicated in the etiology of KS/IHH—ANOS1, FGFR1, FGF8, and PRORK2—were also found in patients with CPHD [41,43,44]. Therefore, the known and candidate genes responsible for pituitary gland development and CPHD were included in the gene panel used in the study (listed in Methods). Two heterozygous missense changes in the SOX3 and GLI2 genes, associated with holoprosencephaly, were found in KS patients—the first previously unreported in any subject without a mutation in other examined known KS/IHH genes, and the second in patients with a deleterious variant in PROKR2: p.R85H (c.254G>A, Figure 1). In all patients, other pituitary hormone deficiencies, except for hypogonadism, were excluded.
In eight patients from the current study (17.4%), P/LP variants were detected in GNRH1, GHRHR1, GLI2, LHX4, and SOX3. Particularly interesting were pathogenic variants in SOX3: p.R155Afs*26 (patient with hyposmia and ao-IHH, proband 34) and combined likely pathogenic variants in LHX4: pG305W/CHD7: p.K850Q in an anosmic patient with absent puberty (proband 8) (Table 1 and Table 2). Novel LP variants in the transcription factor SOX3 (SOX3 p.A234_240del), implicated in the etiology of septo–optic dysplasia (SOD), was observed in an nIHH patient by Kim et al. [45]. Loss-of-function mutations in another transcription factor of the SOX family, SOX10, were linked to KS associated with deafness [46,47,48,49]. Interestingly, heterozygous benign variants in LHX4: p.D128 = were recurring in four patients, of which three turned out to be oligogenic (proband 11, pathogenic variant in CHD7: p.N1030 and benign in HS6ST1: p.K67*; proband 13, pathogenic change in CHD7: p.D2838Tfs*51; and proband 39, benign CHD7 variant p.S103T). Patients with defects in genes responsible for CPHD need careful follow-up and regular evaluation for hypofunction in other pituitary axes.
Two novel P/LP variants in the GNRHR gene were also found (p.C114* and p.N10_Q11delinsKK), both in compound heterozygosity patterns with reported GNRHR defects (probands 24 and 25).
The VUS variant was found in the LRRIQ3 gene (Leucine-Rich Repeats and IQ Motif-Containing 3; LRRC44), a candidate gene for KS/IHH; its expression has been proven in the pituitary gland and the testicles, and its product is probably an intracellular protein [50]. LRRIQ3 is linked with neurodevelopmental disorders and delayed puberty [51,52]. In the current study, the splice-site mutation in LRRIQ3 was found in a 24-year-old patient with severe reproductive phenotype and hyposmia. The possible impact of LRRIQ3 mutations on the development of KS/IHH should be further studied.
4.4. Genotype–Phenotype Correlations
KS patients displayed variable reproductive and non-reproductive phenotypes. Multiple phenotype representations of KS pose significant difficulties in establishing genotype–phenotype correlations.
The detection rate of P/LP variants of selected CPHD/IHH genes was higher in patients with severe non-reproductive phenotypes. Anosmia and additional congenital malformations appear to increase the chance of detecting a casual mutation, especially in GnRH neuron development and migration genes. P/LP variants were detected in patients presenting with anosmia/hyposmia in genes previously reported for nIHH (GNRHR) or SOD/CPHD (LHX4, SOX3). The coexistence of congenital anosmia (whose incidence is < 1/1,000,000) or another cause of smell loss and IHH cannot be ruled out [53,54]. Perhaps a specific sort of defect in GNRHR, LHX4, or SOX3 can result in a complete clinical picture of Kallmann syndrome with smell disturbances. Further studies are required to explore this field.
A reversal of hypogonadism was seen in three of the studied patients (6.5%), slightly less often than expected (10–20%) according to the literature [14,55]. All of the subjects had a P/LP variant in CHD7, in two cases accompanied by P/LP variants in FGFR1 (FGFR1: p.R285W) (unrelated patients 9 and 10). In the third case, a compound defect in GNRHR was associated with a mutation in CHD7 (proband 25). Goncalves et al. described (similar to proband 4) a case of a male patient presenting with partial puberty and nIHH with a reversal in follow-up, diagnosed with a trigenic mutation (FGFR1: c.12G>T; CHD7: c.3245C>T; PROKR2: c.802C>T). Latinen et al., observed reversal in two patients with the same GNRHR mutation (p.R262Q), found also in the current study, which was accompanied by another GNRHR mutation (p.R139H or p. 309delF) and mutations in CHD7 (p.Q51X) or FGFR1 (c.91+2T>A) [56]. Patients with IHH/KS and defects in CHD7 should be significantly monitored for a reversal of hypogonadism. According to the literature, the reversal of IHH is linked with mutations in ANOS1, CHD7, FGFR1, FGF8, GNRHR, HS6ST1, KISS1, KISS1R, NSMF, PROK2, PROKR2, TAC3, and TACR3, and such a minimum panel of 13 genes is recommended in diagnostics of reversal of IHH/KS [24,56,57,58].
The current study’s 13% rate of ao-IHH was comparable with the previously reported 10% incidence of ao-IHH in other IHH/KS study groups [12,59]. Subjects with ao-IHH seem to be more often hyposmic than anosmic, without additional congenital malformations or history of cryptorchidism. The heterogeneity of variants identified in ao-KS patients does not allow for the binding of this form of hypogonadism with a particular genetic defect in the examined population. Ao-IHH in the literature is linked with pathogenic changes in CHD7, FGFR1, FGF8, FGF17, GNRHR, GNRH1, HS6ST1, NROB1, NSMF, PROK2, PROKR2, and WDR11 [14,25,34]. This is the first observation of ao-IHH in a hyposmic patient with a hemizygous pathogenic truncating variant (SOX3: p.R155Afs*26). The significance of SOX3 mutations in the clinical picture of KS/IHH should be studied further.
The molecular diagnostics of KS are today effective and achievable. Costa-Barbosa et al. proposed targeting genetic diagnostics in KS regarding criteria relying on clinical phenotypes [15]. Detailed clinical evaluation of patients and searching for features of concomitant congenital malformations, differentiation of ao-IHH, and observation for reversal forms, are indispensable. Prioritizing genetic testing in KS patients is still challenging regarding the broad heterogeneity of detected defects, as also confirmed in the current study. Interestingly, patients with limb malformation presented mutations in ANOS1, FGFR1, FGF8, or HESX1, which is contrary to previous observations [15,60,61]. The presence of limb anomalies and any other major or additional CHARGE features enables the targeting of molecular diagnostics and testing for the CHD7 gene mutation as the first choice. Limb anomalies are often described as part of the clinical picture of CHD7 mutation and CHARGE syndrome [62]. In patients with synkinesia, searching for mutations in ANOS1, FGFR1/FGF8, and PROK2/PROKR2 should be advised [4,15,60]. However, the presence of mirror movements did not exclude oligogenic mutations in GNRHR and WDR11 (proband 11: GNRHR: p.Q106R and GNRHR p.Ser151 = and WDR11: p.I716V). Renal abnormalities are linked to KS and mutations in ANOS1, CHD7, FGFR1, and FGF8 [4,34,63]. P/LP variants were also reported in ANOS1, CHD7, and FGFR1, as well as GNRHR in patients with renal phenotypes. In case of a lack of additional congenital malformation, panel-based NGS containing at least 15 of the most commonly altered genes (ANOS1, CHD7, FGFR1, FGF8, GNRHR, GNRH1, HS6ST1, KISS1, KISS1R, NSMF, PROK2, PROKR2, TAC3, TACR3, and WDR11) should be used in routine KS diagnostics.
4.5. Oligogenicity
Oligogenic, clinically relevant variants (P/LP) were seen in 11% of studied patients. The achieved diagnostic rate of oligogenicity was comparable to other IHH studies. However, the oligogenicity levels in KS/IHH may differ in various populations. Nair et al. described a 1.5% incidence of oligogenicity in Indians with IHH from Asia [11]. Sykiotis et al. used a panel of eight genes (FGFR1, ANOS1, PROKR2, GNRHR, FGF8, KISS1R, and PROK2) and found digenic mutations in 2% of all studied patients, and 11% of patients with a previously reported monogenic mutation [13]. Quayanor et al. found that the prevalence of digenic mutations detected with an NGS panel of 13 genes (ANOS1, GNRHR, FGFR1, KISS1R, TAC3, TACR3, FGF8, PROKR2, PROK2, CHD7, NSMF, GNRH1, and WDR11) was at the level of 12% [64]. As expected, digenic mutations are more frequent (11–16%) than trigenic ones (about 2% of patients) [13,14,31,58,64,65,66]. Currently, oligogenicity in IHH is considered to affect up to 10–20% of patients [14].
The set of 25 genes—i.e., ANOS1, CHD7, DCC, DUSP6, FGFR1, FGF8, FGF17, FLRT3, GNRHR, HS6ST1, IL17RD, KISS1R, NSMF, NTN1, OL14RD, PNPL46, PROK2, PROKR2, SEMA3A, SEMA7A, SRA1, SPRY, TAC3, TACR3, and WDR11 have been described as inherited in the oligogenic IHH model. [25,67,68,69]. As far as is known, this is the first report evidencing digenic mutations in patients with KS and P/LP variants in PROKR2 and GLI2 (PROKR2: p.R85H and GLI2: p.A185S) genes (proband 30 with anosmia, absence of puberty, and micropenis).
The usage of tests employing analysis of a broad targeted panel of genes significantly raised the probabilityy of detection for clinically relevant changes in CPHD/IHH genes. Considering variants of all classes of pathogenicity, the prevalence of oligogenic variants was up to the level of 32.6%. The ACMG’s classification scheme is primarily intended to classify variants in Mendelian diseases’ genes, and was developed for the evaluation of variants in monogenic disorders [70].
For this reason, variants in all ACMG classes in IHH/CPHD have been reported in the current study. VUS alterations represent an unclear portion of defects, but considering their low frequency and in silico predictions, they could impair protein function, and are hypothesized to contribute to an oligogenic etiology and the development of the disease phenotype [71,72]. There are a growing number of studies, including VUS variants, investigating the possibility of providing a molecular diagnosis in case of reinterpretation of variants [73]. Future studies are needed in order to determine the pathogenicity of VUS variants to IHH/KS etiology.
The impact of the coexistence of different variants in a few genes on phenotype is still difficult to predict [13,14,16]. Two different genetic defects may have a synergistic effect [74]. In familial IHH cases, mutations in different loci give rise to slightly different phenotypes [65]. Synergistic heterozygosity, assuming the existence of several partial protein defects in at least one signaling pathway, has been thoroughly studied in cardiac and metabolic diseases and cancers [75,76,77]. Synergistic heterozygosity models in KS/IHH have been proposed for the mutations in NSMF and ANOS1; NSMF and TACR3; FGF8, FGFR1, and ANOS1 [66,78]. A modifying, synergistic effect cannot be excluded regarding the coexistence of the P/LP, VUS, and perhaps even B/LB variants in critical genes essential for neuronal migration, the development of the pituitary gland, or signal transduction. The incidence of detected oligogenicity of KS in various studies relies significantly on the applied molecular method, assuming the number of analyzed genes, the variant classification criteria applied and, finally, the size of the examined cohort. There is a need for further research on KS/IHH using a comprehensive NGS approach with a wide panel of genes, whole-exome sequencing (WES), or even attempting patient whole-genome sequencing (WGS) if previous strategies fail to find a causative defect, in order to precisely define phenotypes [79].
The possible outcome of accumulated variants, principally P/LP and VUS, in more than one IHH/CPHD gene in the same patient should be continually studied. The significance of synergistic heterozygosity in IHH/KS pathogenesis represents a great challenge for further research.
5. Conclusions
In conclusion, this study has identified 20 new P/LP mutations in ANOS1, CHD7, FGFR1, GLI2, GNRHR, SOX3, and WDR11. The molecular basis of KS has been established for 43.5% of all studied patients. KS patients originating from Poland displayed variable reproductive and non-reproductive phenotypes, not always corresponding with previous data on phenotype–genotype correlation. The use of an NGS strategy employing a comprehensive panel of genes increases the chances of detecting monogenetic and oligogenic defects in KS patients presenting typical clinical characteristics. Mutations in genes responsible for GnRH neuron migration and development constitute the most frequent defects in studied subjects. The absence of recurrent alterations confirms the high heterogeneity of mutations, as evidenced in other studies. The relatively high incidence of oligogenicity in KS represents a diagnostic and interpretational challenge. The impact of oligogenicity and the relatively high VUS incidence on KS phenotype and disease course requires further observation. The role of CPHD genes, especially LHX4 and SOX3, in KS development should be also further studied.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes12060868/s1, Table S1: Variants Information.
Author Contributions
Conceptualization, M.K., B.B., and K.Z.; Methodology, B.B. and M.K.; software, B.B.; Validation, B.B. and M.K.; Formal analysis, M.K., B.B., and K.Z.; Investigation, M.K., E.W., M.R., J.K., A.D., M.T.-M., and A.H.-D.; Resources, M.K.; Data curation, M.K.; Writing—original draft preparation, M.K., B.B., and K.Z.; Writing—review and editing, M.K., B.B., and K.Z.; Visualization, M.R.(Michał Rabijewski); Supervision, K.Z. and M.R. (Marek Ruchała).; project administration, M.K. and B.B.; Funding acquisition, M.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was conducted according to research project No. 2016/21/N/NZ5/00511, funded by the National Science Center. The APC was funded by National Science Center and Poznan University of Medical Science.
Institutional Review Board Statement
This study was conducted in accordance with the guidelines of the Declaration of Helsinki, and approved by the Institutional Ethics Committee of Poznan University of Medical Science (1002/13, 5 December 2013; 990/15, 5 November 2015, 567/16, 5 May 2016).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors would like to thank the patients for their contribution to the study, and the employees of the Clinical Department of Phoniatrics and Audiology, PUMS, for performing the olfactory test on KS patients.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bianco, S.D.C.; Kaiser, U.B. The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism. Nat. Rev. Endocrinol. 2009, 5, 569–576. [Google Scholar] [CrossRef]
- Schwanzel-Fukuda, M.; Pfaff, D.W. Origin of luteinizing hormone-releasing hormone neurons. Nat. Cell Biol. 1989, 338, 161–164. [Google Scholar] [CrossRef]
- Wray, S. Development of gonadotropin-releasing hormone-1 neurons. Front. Neuroendocr. 2002, 23, 292–316. [Google Scholar] [CrossRef]
- Tickotsky, N.; Moskovitz, M. Renal Agenesis in Kallmann Syndrome: A Network Approach. Ann. Hum. Genet. 2014, 78, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Quinton, R.; Duke, V.M.; Robertson, A.; Kirk, J.M.W.; Matfin, G.; De Zoysa, P.A.; Azcona, C.; MacColl, G.S.; Jacobs, H.S.; Conway, G.S.; et al. Idiopathic gonadotrophin deficiency: Genetic questions addressed through phenotypic characterization. Clin. Endocrinol. 2001, 55, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Della Valle, E.; Vezzani, S.; Rochira, V.; Granata, A.R.M.; Madeo, B.; Genovese, E.; Pignatti, E.; Marino, M.; Carani, C.; Simoni, M. Prevalence of Olfactory and Other Developmental Anomalies in Patients with Central Hypogonadotropic Hypogonadism. Front. Endocrinol. 2013, 4, 70. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, H.-G.; Kurth, I.; Lan, F.; Meliciani, I.; Wenzel, W.; Eom, S.H.; Kang, G.B.; Rosenberger, G.; Tekin, M.; Ozata, M.; et al. Mutations in CHD7, Encoding a Chromatin-Remodeling Protein, Cause Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Am. J. Hum. Genet. 2008, 83, 511–519. [Google Scholar] [CrossRef]
- Pinto, G.; Abadie, V.; Mesnage, R.; Blustajn, J.; Cabrol, S.; Amiel, J.; Hertz-Pannier, L.; Bertrand, A.M.; Lyonnet, S.; Rappaport, R.; et al. Charge Syndrome Includes Hypogonadotropic Hypogonadism and Abnormal Olfactory Bulb Development. J. Clin. Endocrinol. Metab. 2005, 90, 5621–5626. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.-J.; Shan, Y.; Whittington, N.C.; Wray, S. Nasal Placode Development, GnRH Neuronal Migration and Kallmann Syndrome. Front. Cell Dev. Biol. 2019, 7, 121. [Google Scholar] [CrossRef]
- Laitinen, E.-M.; Vaaralahti, K.; Tommiska, J.; Eklund, E.; Tervaniemi, M.; Valanne, L.; Raivio, T. Incidence, Phenotypic Features and Molecular Genetics of Kallmann Syndrome in Finland. Orphanet J. Rare Dis. 2011, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Jadhav, S.; Lila, A.; Jagtap, V.; Bukan, A.; Pandit, R.; Ekbote, A.; Dharmalingam, M.; Kumar, P.; Kalra, P.; et al. Spectrum of phenotype and genotype of congenital isolated hypogonadotropic hypogonadism in Asian Indians. Clin. Endocrinol. 2016, 85, 100–109. [Google Scholar] [CrossRef]
- Bonomi, M.; Vezzoli, V.; Krausz, C.; Guizzardi, F.; Vezzani, S.; Simoni, M.; Bassi, I.; Duminuco, P.; di Iorgi, N.; Giavoli, C.; et al. Characteristics of a Nationwide Cohort of Patients Presenting with Isolated Hy-pogonadotropic Hypogonadism (Ihh). Eur. J. Endocrinol. 2018, 178, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Sykiotis, G.P.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.A.; Quinton, R.; Hall, J.E.; Gusella, J.F.; et al. Oligogenic Basis of Isolated Gonadotropin-Releasing Hormone Deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 15140–15144. [Google Scholar] [CrossRef]
- Topaloğlu, A.K. Update on the Genetics of Idiopathic Hypogonadotropic Hypogonadism. J. Clin. Res. Pediatr. Endocrinol. 2018, 9, 113–122. [Google Scholar] [CrossRef]
- Costa-Barbosa, F.A.; Balasubramanian, R.; Keefe, K.W.; Shaw, N.; Al Tassan, N.; Plummer, L.; Dwyer, A.A.; Buck, C.L.; Choi, J.-H.; Seminara, S.B.; et al. Prioritizing Genetic Testing in Patients with Kallmann Syndrome Using Clinical Phenotypes. J. Clin. Endocrinol. Metab. 2013, 98, E943–E953. [Google Scholar] [CrossRef] [PubMed]
- Maione, L.; Dwyer, A.A.; Francou, B.; Guiochon-Mantel, A.; Binart, N.; Bouligand, J.; Young, J. Genetics in Endocrinology: Genetic counseling for congenital hypogonadotropic hypogonadism and Kallmann syndrome: New challenges in the era of oligogenism and next-generation sequencing. Eur. J. Endocrinol. 2018, 178, R55–R80. [Google Scholar] [CrossRef]
- Rojek, A.; Obara-Moszynska, M.; Malecka, E.; Slomko-Jozwiak, M.; Niedziela, M. NR0B1 (DAX1) mutations in patients affected by congenital adrenal hypoplasia with growth hormone deficiency as a new finding. J. Appl. Genet. 2013, 54, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Dzaman, K.; Zborowska-Piskadlo, K.; Pietniczka-Zaleska, M.; Kantor, I. Kallmann Syndrome in Pediatric Otorhinolar-yngology Practice—Case Report and Literature Review. Int. J. Pediatr. Otorhinolaryngol. 2017, 100, 149–153. [Google Scholar] [CrossRef] [PubMed]
- Placzkiewicz, E.; Baldys-Waligorska, A. Kallmann’s Syndrome: Skeletal and Psychological Aspects of Late Diagnosis. Ann. Endocrinol. 2003, 64, 277–280. [Google Scholar]
- Noczynska, A.; Wasikowa, R. Kallmann’s Syndrome in Families. Endokrynol. Diabetol. Chor. Przemiany Mater. Wieku Rozw. 2004, 10, 127–131. [Google Scholar]
- Pierzchlewska, M.M.; Robaczyk, M.G.; Vogel, I. Induction of puberty with human chorionic gonadotropin (hCG) followed by reversal of hypogonadotropic hypogonadism in Kallmann syndrome. Endokrynol. Polska 2015, 68, 692–696. [Google Scholar] [CrossRef]
- Gach, A.; Pinkier, I.; Sałacińska, K.; Szarras-Czapnik, M.; Salachna, D.; Kucińska, A.; Rybak-Krzyszkowska, M.; Sakowicz, A. Identification of gene variants in a cohort of hypogonadotropic hypogonadism: Diagnostic utility of custom NGS panel and WES in unravelling genetic complexity of the disease. Mol. Cell. Endocrinol. 2020, 517, 110968. [Google Scholar] [CrossRef]
- Gach, A.; Pinkier, I.; Szarras-Czapnik, M.; Sakowicz, A.; Jakubowski, L. Expanding the mutational spectrum of monogenic hypogonadotropic hypogonadism: Novel mutations in ANOS1 and FGFR1 genes. Reprod. Biol. Endocrinol. 2020, 18, 8. [Google Scholar] [CrossRef]
- Mao, J.F.; Xu, H.L.; Duan, J.; Chen, R.R.; Li, L.; Li, B.; Nie, M.; Min, L.; Zhang, H.B.; Wu, X.Y. Reversal of Idiopathic Hy-pogonadotropic Hypogonadism: A Cohort Study in Chinese Patients. Asian J. Androl. 2015, 17, 497–502. [Google Scholar]
- Boehm, U.; Bouloux, P.M.; Dattani, M.T.; de Roux, N.; Dode, C.; Dunkel, L.; Dwyer, A.A.; Giacobini, P.; Hardelin, J.P.; Juul, A.; et al. Expert Consensus Document: Eu-ropean Consensus Statement on Congenital Hypogonadotropic Hypogonadism—Pathogenesis, Diagnosis and Treatment. Nat. Rev. Endocrinol. 2015, 11, 547–564. [Google Scholar] [CrossRef]
- Elsberg, C.A.; Levy, I.; Brewer, E.D.; Merrill, M.H.; Lacaillade, C.W.; Broeck, C.T. A new method for testing the sense of SMELL and for the establishment of olfactory values of odorous substances. Science 1936, 83, 211–212. [Google Scholar] [CrossRef]
- Pruszewicz, A. Apropos of Gustatory and Olfactory Tests. Otolaryngol. Polska 1965, 19, 29–37. [Google Scholar]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Lasker, K.; Schneidman-Duhovny, D.; Webb, B.; Huang, C.C.; Pettersen, E.F.; Goddard, T.D.; Meng, E.C.; Sali, A.; Ferrin, T.E. UCSF Chimera, MODELLER, and IMP: An integrated modeling system. J. Struct. Biol. 2012, 179, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Quaynor, S.D.; Bosley, M.E.; Duckworth, C.G.; Porter, K.R.; Kim, S.-H.; Kim, H.-G.; Chorich, L.P.; Sullivan, M.E.; Choi, J.-H.; Cameron, R.S.; et al. Targeted next generation sequencing approach identifies eighteen new candidate genes in normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Cell. Endocrinol. 2016, 437, 86–96. [Google Scholar] [CrossRef]
- Butz, H.; Nyírő, G.; Kurucz, P.A.; Likó, I.; Patócs, A. Molecular genetic diagnostics of hypogonadotropic hypogonadism: From panel design towards result interpretation in clinical practice. Qual. Life Res. 2021, 140, 113–134. [Google Scholar] [CrossRef]
- Cangiano, B.; Duminuco, P.; Vezzoli, V.; Guizzardi, F.; Chiodini, I.; Corona, G.; Maggi, M.; Persani, L.; Bonomi, M. Evidence for a Common Genetic Origin of Classic and Milder Adult-Onset Forms of Isolated Hypogonadotropic Hypogonadism. J. Clin. Med. 2019, 8, 126. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Crowle, W. Isolated Gonadotropin-Releasing Hormone (Gnrh) Deficiency; University of Washington: Seattle, WA, USA, 2007. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1334/ (accessed on 23 May 2021).
- Xu, C.; Cassatella, D.; van der Sloot, A.M.; Quinton, R.; Hauschild, M.; de Geyter, C.; Fluck, C.; Feller, K.; Bartholdi, D.; Nemeth, A.; et al. Evaluating Charge Syndrome in Congenital Hypogonadotropic Hypogonadism Patients Harboring Chd7 Variants. Genet. Med. 2018, 20, 872–881. [Google Scholar] [CrossRef]
- Kim, H.G.; Layman, L.C. The Role of Chd7 and the Newly Identified Wdr11 Gene in Patients with Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Mol. Cell Endocrinol. 2011, 346, 74–83. [Google Scholar] [CrossRef]
- Turan, I.; Hutchins, B.I.; Hacihamdioglu, B.; Kotan, L.D.; Gurbuz, F.; Ulubay, A.; Mengen, E.; Yuksel, B.; Wray, S.; Topaloglu, A.K. CCDC141 Mutations in Idiopathic Hypogonadotropic Hypogonadism. J. Clin. Endocrinol. Metab. 2017, 102, 1816–1825. [Google Scholar] [CrossRef] [PubMed]
- Tusset, C.; Trarbach, E.B.; Silveira, L.F.; Beneduzzi, D.; Montenegro, L.; Latronico, A.C. Clinical and Molecular Aspects of Congenital Isolated Hypogonadotropic Hypogonadism. Arq. Bras. Endocrinol. Metabol. 2011, 55, 501–511. [Google Scholar] [CrossRef][Green Version]
- Dode, C.; Teixeira, L.; Levilliers, J.; Fouveaut, C.; Bouchard, P.; Kottler, M.L.; Lespinasse, J.; Lienhardt-Roussie, A.; Mathieu, M.; Moerman, A.; et al. Kallmann Syndrome: Mutations in the Genes Encoding Prokineticin-2 and Prokineticin Receptor-2. PLoS Genet. 2006, 2, e175. [Google Scholar] [CrossRef] [PubMed]
- Monnier, C.; Dodé, C.; Fabre, L.; Teixeira, L.; Labesse, G.; Pin, J.-P.; Hardelin, J.-P.; Rondard, P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum. Mol. Genet. 2008, 18, 75–81. [Google Scholar] [CrossRef]
- Raivio, T.; Avbelj, M.; McCabe, M.J.; Romero, C.J.; Dwyer, A.A.; Tommiska, J.; Sykiotis, G.P.; Gregory, L.C.; Diaczok, D.; Tziaferi, V.; et al. Genetic Overlap in Kallmann Syndrome, Combined Pituitary Hormone Deficiency, and Septo-Optic Dysplasia. J. Clin. Endocrinol. Metab. 2012, 97, E694–E699. [Google Scholar] [CrossRef] [PubMed]
- Vaaralahti, K.; Raivio, T.; Koivu, R.; Valanne, L.; Laitinen, E.-M.; Tommiska, J. Genetic Overlap between Holoprosencephaly and Kallmann Syndrome. Mol. Syndr. 2012, 3, 1–5. [Google Scholar] [CrossRef]
- Asakura, Y.; Muroya, K.; Hanakawa, J.; Sato, T.; Aida, N.; Narumi, S.; Hasegawa, T.; Adachi, M. Combined Pituitary Hormone Deficiency with Unique Pituitary Dysplasia and Morning Glory Syndrome Related to a Heterozygous Prokr2 Mutation. Clin. Pediatr. Endocrinol. 2015, 24, 27–32. [Google Scholar] [CrossRef]
- Correa, F.A.; Trarbach, E.B.; Tusset, C.; Latronico, A.C.; Montenegro, L.R.; Carvalho, L.R.; Franca, M.M.; Otto, A.P.; Costalonga, E.F.; Brito, V.N.; et al. FGFR1 and PROKR2 rare variants found in patients with combined pituitary hormone deficiencies. Endocr. Connect. 2015, 4, 100–107. [Google Scholar] [CrossRef]
- Kim, J.H.; Seo, G.H.; Kim, G.-H.; Huh, J.; Hwang, I.T.; Jang, J.-H.; Yoo, H.-W.; Choi, J.-H. Targeted Gene Panel Sequencing for Molecular Diagnosis of Kallmann Syndrome and Normosmic Idiopathic Hypogonadotropic Hypogonadism. Exp. Clin. Endocrinol. Diabetes 2018, 127, 538–544. [Google Scholar] [CrossRef]
- Vaaralahti, K.; Tommiska, J.; Tillmann, V.; Liivak, N.; Känsäkoski, J.; Laitinen, E.-M.; Raivio, T. De novo SOX10 nonsense mutation in a patient with Kallmann syndrome and hearing loss. Pediatr. Res. 2014, 76, 115–116. [Google Scholar] [CrossRef][Green Version]
- Pingault, V.; Bodereau, V.; Baral, V.; Marcos, S.; Watanabe, Y.; Chaoui, A.; Fouveaut, C.; Leroy, C.; Vérier-Mine, O.; Francannet, C.; et al. Loss-of-Function Mutations in SOX10 Cause Kallmann Syndrome with Deafness. Am. J. Hum. Genet. 2013, 92, 707–724. [Google Scholar] [CrossRef]
- Suzuki, E.; Izumi, Y.; Chiba, Y.; Horikawa, R.; Matsubara, Y.; Tanaka, M.; Ogata, T.; Fukami, M.; Naiki, Y. Loss-of-Function SOX10 Mutation in a Patient with Kallmann Syndrome, Hearing Loss, and Iris Hypopigmentation. Horm. Res. Paediatr. 2015, 84, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zhao, S.; Xie, Y.; Yang, W.; Mo, Z. De novo SOX10 Nonsense Mutation in a Patient with Kallmann Syndrome, Deafness, Iris Hypopigmentation, and Hyperthyroidism. Ann. Clin. Lab. Sci. 2018, 48, 248–252. [Google Scholar] [PubMed]
- Lrriq3. Available online: https://www.proteinatlas.org/ENSG00000162620-LRRIQ3/tissue (accessed on 1 April 2021).
- Reuter, M.S.; Tawamie, H.; Buchert, R.; Gebril, O.H.; Froukh, T.; Thiel, C.; Uebe, S.; Ekici, A.B.; Krumbiegel, M.; Zweier, C.; et al. Diagnostic Yield and Novel Candidate Genes by Exome Sequencing in 152 Consanguineous Families with Neurodevelopmental Disorders. JAMA Psychiatry 2017, 74, 293–299. [Google Scholar] [CrossRef]
- Howard, S.R.; Guasti, L.; Ruiz-Babot, G.; Mancini, A.; David, A.; Storr, H.L.; Metherell, L.A.; Sternberg, M.J.; Cabrera, C.P.; Warren, H.R.; et al. Igsf10 Mutations Dysregulate Gonadotropin-Releasing Hormone Neuronal Migration Resulting in Delayed Puberty. EMBO Mol. Med. 2016, 8, 626–642. [Google Scholar] [CrossRef] [PubMed]
- Karstensen, H.; Tommerup, N. Isolated and syndromic forms of congenital anosmia. Clin. Genet. 2011, 81, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Dalton, P.; Mennella, J.A.; Cowart, B.J.; Maute, C.; Pribitkin, E.A.; Reilly, J.S. Evaluating the Prevalence of Olfactory Dysfunction in a Pediatric Population. Ann. N. Y. Acad. Sci. 2009, 1170, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Raivio, T.; Falardeau, J.; Dwyer, A.; Quinton, R.; Hayes, F.J.; Hughes, V.A.; Cole, L.W.; Pearce, S.H.; Lee, H.; Boepple, P.; et al. Reversal of Idiopathic Hypogonadotropic Hypogonadism. N. Engl. J. Med. 2007, 357, 863–873. [Google Scholar] [CrossRef]
- Laitinen, E.-M.; Tommiska, J.; Sane, T.; Vaaralahti, K.; Toppari, J.; Raivio, T. Reversible Congenital Hypogonadotropic Hypogonadism in Patients with CHD7, FGFR1 or GNRHR Mutations. PLoS ONE 2012, 7, e39450. [Google Scholar] [CrossRef]
- Bouligand, J.; Ghervan, C.; Tello, J.A.; Brailly-Tabard, S.; Salenave, S.; Chanson, P.; Lombès, M.; Millar, R.P.; Guiochon-Mantel, A.; Young, J. Isolated Familial Hypogonadotropic Hypogonadism and aGNRH1Mutation. N. Engl. J. Med. 2009, 360, 2742–2748. [Google Scholar] [CrossRef]
- Cole, L.W.; Sidis, Y.; Zhang, C.; Quinton, R.; Plummer, L.; Pignatelli, D.; Hughes, V.A.; Dwyer, A.A.; Raivio, T.; Hayes, F.J.; et al. Mutations in Prokineticin 2andProkineticin receptor 2genes in Human Gonadotrophin-Releasing Hormone Deficiency: Molecular Genetics and Clinical Spectrum. J. Clin. Endocrinol. Metab. 2008, 93, 3551–3559. [Google Scholar] [CrossRef]
- Nachtigall, L.B.; Boepple, P.A.; Pralong, F.P.; Crowley, W.F., Jr. Adult-Onset Idiopathic Hypogonadotropic Hypogonadism—A Treatable Form of Male Infertility. N. Engl. J. Med. 1997, 336, 410–415. [Google Scholar] [CrossRef]
- Knickmeyer, R.C.; Auyeung, B.; Davenport, M.L. Assessing Prenatal and Neonatal Gonadal Steroid Exposure for Studies of Human Development: Methodological and Theoretical Challenges. Front. Endocrinol. 2015, 5, 242. [Google Scholar] [CrossRef] [PubMed]
- Trarbach, E.B.; Abreu, A.P.; Silveira, L.F.; Garmes, H.M.; Baptista, M.T.; Teles, M.G.; Costa, E.M.; Mohammadi, M.; Pitteloud, N.; Mendonca, B.B.; et al. Nonsense Mutations in Fgf8 Gene Causing Different Degrees of Human Gonadotropin-Releasing Deficiency. J. Clin. Endocrinol. Metab. 2010, 95, 3491–3496. [Google Scholar] [CrossRef] [PubMed]
- Brock, K.E.; Mathiason, M.A.; Rooney, B.L.; Williams, M.S. Quantitative analysis of limb anomalies in CHARGE syndrome: Correlation with diagnosis and characteristic CHARGE anomalies. Am. J. Med. Genet. Part A 2009, 123, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Sanna-Cherchi, S.; Caridi, G.; Weng, P.L.; Scolari, F.; Perfumo, F.; Gharavi, A.G.; Ghiggeri, G.M. Genetic Approaches to Human Renal Agenesis/Hypoplasia and Dysplasia. Pediatr. Nephrol. 2007, 22, 1675–1684. [Google Scholar] [CrossRef] [PubMed]
- Quaynor, S.D.; Kim, H.G.; Cappello, E.M.; Williams, T.; Chorich, L.P.; Bick, D.P.; Sherins, R.J.; Layman, L.C. The Prevalence of Digenic Mutations in Patients with Normosmic Hypogonadotropic Hypogonadism and Kallmann Syndrome. Fertil. Steril. 2011, 96, 1424–1430.e6. [Google Scholar] [CrossRef]
- Pitteloud, N.; Quinton, R.; Pearce, S.; Raivio, T.; Acierno, J.; Dwyer, A.; Plummer, L.; Hughes, V.; Seminara, S.; Cheng, Y.-Z.; et al. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Investig. 2007, 117, 457–463. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Kim, H.-G.; Bhagavath, B.; Cho, S.-G.; Lee, J.H.; Ha, K.; Meliciani, I.; Wenzel, W.; Podolsky, R.H.; Chorich, L.P.; et al. Nasal embryonic LHRH factor (NELF) mutations in patients with normosmic hypogonadotropic hypogonadism and Kallmann syndrome. Fertil. Steril. 2011, 95, 1613–1620.e7. [Google Scholar] [CrossRef]
- Miraoui, H.; Dwyer, A.A.; Sykiotis, G.P.; Plummer, L.; Chung, W.; Feng, B.; Beenken, A.; Clarke, J.; Pers, T.H.; Dworzynski, P.; et al. Mutations in Fgf17, Il17rd, Dusp6, Spry4, and Flrt3 Are Identified in Individuals with Congenital Hypogonadotropic Hy-pogonadism. Am. J. Hum. Genet. 2013, 92, 725–743. [Google Scholar] [CrossRef] [PubMed]
- Bouilly, J.; Messina, A.; Papadakis, G.; Cassatella, D.; Xu, C.; Acierno, J.S.; Tata, B.; Sykiotis, G.; Santini, S.; Sidis, Y.; et al. Dcc/Ntn1 Complex Mutations in Patients with Congenital Hypogonadotropic Hypogonadism Impair Gnrh Neuron Development. Hum. Mol. Genet. 2018, 27, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Kotan, L.D.; Hutchins, B.I.; Ozkan, Y.; Demirel, F.; Stoner, H.; Cheng-Hathaway, P.; Esen, I.; Gurbuz, F.; Bicakci, Y.K.; Mengen, E.; et al. Mutations in FEZF1 Cause Kallmann Syndrome. Am. J. Hum. Genet. 2014, 95, 326–331. [Google Scholar] [CrossRef]
- Amendola, L.M.; Jarvik, G.P.; Leo, M.C.; McLaughlin, H.M.; Akkari, Y.; Amaral, M.D.; Berg, J.S.; Biswas, S.; Bowling, K.M.; Conlin, L.K.; et al. Performance of ACMG-AMP Variant-Interpretation Guidelines among Nine Laboratories in the Clinical Sequencing Exploratory Research Consortium. Am. J. Hum. Genet. 2016, 98, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Szot, J.; Cuny, H.; Blue, G.M.; Humphreys, D.T.; Ip, E.; Harrison, K.; Sholler, G.F.; Giannoulatou, E.; Leo, P.; Duncan, E.L.; et al. A Screening Approach to Identify Clinically Actionable Variants Causing Congenital Heart Disease in Exome Data. Circ. Genom. Precis. Med. 2018, 11, e001978. [Google Scholar] [CrossRef]
- Alosi, D.; Bisgaard, M.L.; Hemmingsen, S.N.; Krogh, L.N.; Mikkelsen, H.B.; Binderup, M.L.M. Management of Gene Variants of Unknown Significance: Analysis Method and Risk Assessment of the Vhl Mutation P.P81s (C.241c>T). Curr. Genom. 2017, 18, 93–103. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vears, D.F.; Niemiec, E.; Howard, H.C.; Borry, P. Analysis of VUS reporting, variant reinterpretation and recontact policies in clinical genomic sequencing consent forms. Eur. J. Hum. Genet. 2018, 26, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Pérez, J.M.; Candela, H.; Micol, J.L. Understanding synergy in genetic interactions. Trends Genet. 2009, 25, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Pulignani, S.; Vecoli, C.; Borghini, A.; Foffa, I.; Ait-Ali, L.; Andreassi, M.G. Targeted Next-Generation Sequencing in Patients with Non-syndromic Congenital Heart Disease. Pediatr. Cardiol. 2018, 39, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Cervera-Acedo, C.; Coloma, A.; Huarte-Loza, E.; Sierra-Carpio, M.; Domínguez-Garrido, E. Phenotype variability in a large Spanish family with Alport syndrome associated with novel mutations in COL4A3 gene. BMC Nephrol. 2017, 18, 325. [Google Scholar] [CrossRef]
- Niku, M.; Pajari, A.-M.; Sarantaus, L.; Päivärinta, E.; Storvik, M.; Heiman-Lindh, A.; Suokas, S.; Nyström, M.; Mutanen, M. Western diet enhances intestinal tumorigenesis in Min/+ mice, associating with mucosal metabolic and inflammatory stress and loss of Apc heterozygosity. J. Nutr. Biochem. 2017, 39, 126–133. [Google Scholar] [CrossRef] [PubMed]
- González-Martínez, D.; Kim, S.-H.; Hu, Y.; Guimond, S.; Schofield, J.; Winyard, P.; Vannelli, G.B.; Turnbull, J.; Bouloux, P.-M. Anosmin-1 Modulates Fibroblast Growth Factor Receptor 1 Signaling in Human Gonadotropin-Releasing Hormone Olfactory Neuroblasts through a Heparan Sulfate-Dependent Mechanism. J. Neurosci. 2004, 24, 10384–10392. [Google Scholar] [CrossRef]
- Cassatella, D.; Howard, S.; Acierno, J.S.; Xu, C.; Papadakis, G.E.; Santoni, F.A.; Dwyer, A.A.; Santini, S.; Sykiotis, G.P.; Chambion, C.; et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. Eur. J. Endocrinol. 2018, 178, 377–388. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).