
Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes



Abstract
:1. Introduction
2. Fetal Epigenetic Imprinting and Maternal Factors
3. Non-Imprinting Epigenetic Changes in Prenatal Life
4. Epigenetic Changes of Immune-Related Genes
5. Fetal Epigenetic Changes: Studies on Cord Blood Cells
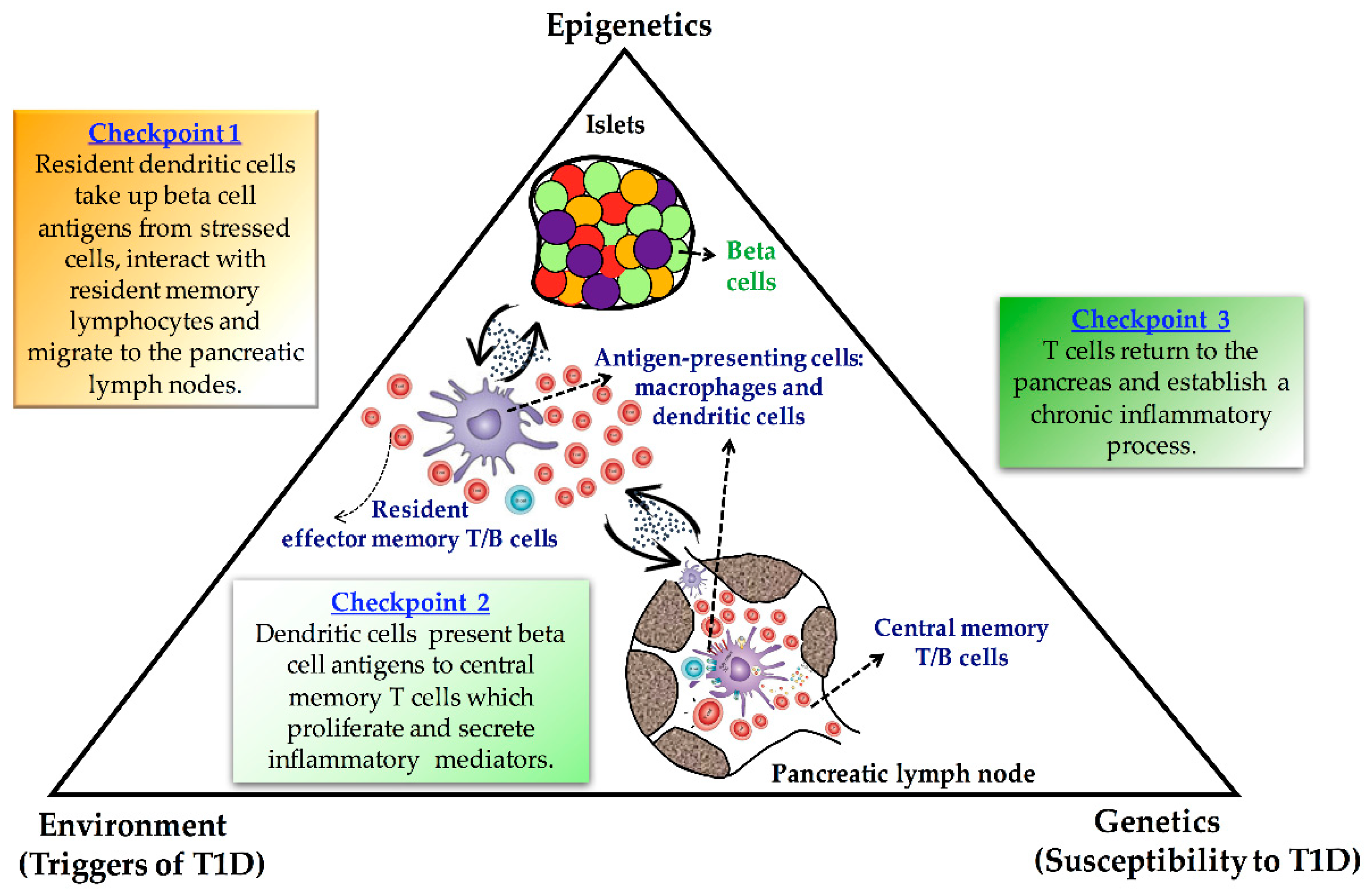
6. Epigenetics in T1D: The Missing Piece of the Puzzle
6.1. Genetics
6.2. Genome Imprinting
6.3. Non-Imprinting Epigenetic Changes
6.4. DNA Methylation Signature in T1D
6.5. Maternal Autoantibodies and Their Role in T1D
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Roep, B.O. The role of T-cells in the pathogenesis of type 1 diabetes: From cause to cure. Diabetologia 2003, 46, 305–321. [Google Scholar] [CrossRef]
- Atkinson, M.A.; Eisenbarth, G.S.; Michels, A.W. Type 1 diabetes. Lancet 2014, 383, 69–82. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Jeffrey, J.; Fain, P.R.; Eisenbarth, G.S.; Orban, T. Concordance for islet autoimmunity among monozygotic twins. N. Engl. J. Med. 2008, 359, 2849–2850. [Google Scholar] [CrossRef] [PubMed]
- Islam, T.; Gauderman, W.J.; Cozen, W.; Hamilton, A.S.; Burnett, M.E.; Mack, T.M. Differential twin concordance for multiple sclerosis by latitude of birthplace. Ann. Neurol. 2006, 60, 56–64. [Google Scholar] [CrossRef]
- Vojdani, A. A Potential link between environmental triggers and autoimmunity. Autoimmune Dis. 2014, 437231. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.F.; Wang, G. Environmental agents, oxidative stress and autoimmunity. Curr. Opin. Toxicol. 2018, 7, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.F.; Wang, H. Environmental exposures and autoimmune diseases: Contribution of gut microbiome. Front. Immunol. 2020, 10, 3094. [Google Scholar] [CrossRef] [PubMed]
- Predieri, B.; Bruzzi, P.; Bigi, E.; Ciancia, S.; Madeo, S.F.; Lucaccioni, L.; Lughetti, L. Endocrine disrupting chemicals and type 1 diabetes. Int. J. Mol. Sci. 2020, 21, 2937. [Google Scholar] [CrossRef] [PubMed]
- Cavalli, G.; Heard, E. Advances in epigenetics link genetics to the environment and disease. Nature 2019, 571, 489–499. [Google Scholar] [CrossRef] [Green Version]
- Gibney, E.; Nolan, C. Epigenetics and gene expression. Heredity 2010, 105, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y. Modern epigenetics methods in biological research. Methods 2021, 187, 104–113. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, M.V.C.; Bourc’his, D. The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 2019, 20, 590–607. [Google Scholar] [CrossRef]
- Ferguson-Smith, A.C.; Bourc’his, D. The discovery and importance of genomic imprinting. eLife 2018, 7, e42368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucci, V.; Isles, A.R.; Kelsey, G.; Ferguson-Smith, A.C.; Erice Imprinting Group. Genomic imprinting and physiological processes in mammals. Cell 2019, 176, 952–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hitchcock, T.J.; Paracchini, S.; Gardner, A. Genomic imprinting as a window into human language evolution. Bioessays 2019, 41, e1800212. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.; Schübeler, D. Genomic patterns of DNA methylation: Targets and function of an epigenetic mark. Curr. Opin. Cell. Biol. 2007, 19, 273–280. [Google Scholar] [CrossRef]
- Miranda, T.B.; Jones, P.A. DNA methylation: The nuts and bolts of repression. J. Cell. Physiol. 2007, 213, 384–390. [Google Scholar] [CrossRef]
- Bartolomei, M.S.; Ferguson-Smith, A.C. Mammalian genomic imprinting. Cold Spring Harb. Perspect. Biol. 2011, 3, a002592. [Google Scholar] [CrossRef] [Green Version]
- Kaneko-Ishino, T.; Ishino, F. Evolution of viviparity in mammals: What genomic imprinting tells us about mammalian placental evolution. Reprod. Fertil. Dev. 2019, 31, 1219–1227. [Google Scholar] [CrossRef]
- Hanna, C.W. Placental imprinting: Emerging mechanisms and functions. PLoS Genet. 2020, 16, e1008709. [Google Scholar] [CrossRef]
- Millership, S.J.; Van de Pette, M.; Withers, D.J. Genomic imprinting and its effects on postnatal growth and adult metabolism. Cell. Mol. Life. Sci. 2019, 76, 4009–4021. [Google Scholar] [CrossRef] [Green Version]
- Trasler, J.M. Gamete imprinting: Setting epigenetic patterns for the next generation. Reprod Fertil. Dev. 2006, 18, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thamban, T.; Agarwaal, V.; Khosla, S. Role of genomic imprinting in mammalian development. J. Biosci. 2020, 45, 20. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, E.; Dolinoy, D.C.; Mancuso, P. Perinatal bisphenol A exposures increase production of pro-inflammatory mediators in bone marrow-derived mast cells of adult mice. Immunotoxicology 2014, 11, 205–212. [Google Scholar] [CrossRef]
- Jahreis, S.; Trump, S.; Bauer, M.; Bauer, T.; Thürmann, L.; Feltens, R.; Wang, Q.; Gu, L.; Grützmann, K.; Röder, S.; et al. Maternal phthalate exposure promotes allergic airway inflammation over 2 generations through epigenetic modifications. J. Allergy Clin. Immunol. 2018, 141, 741–753. [Google Scholar] [CrossRef] [Green Version]
- Ringh, M.V.; Hagemann-Jensen, M.; Needhamsen, M.; Kular, L.; Breeze, C.E.; Sjöholm, L.K.; Slavec, L.; Kullber, S.; Wahlström, J.; Grunewald, J.; et al. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells. EBioMedicine 2019, 46, 290–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, W.; Puttabyatappa, M.; Zeng, L.; Vazquez, D.; Pennathur, S.; Padmanabhan, V. Developmental programming: Prenatal bisphenol A treatment disrupts mediators of placental function in sheep. Chemosphere 2020, 243, 125301. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A.; Beedle, A.S. Non-genomic transgenerational inheritance of disease risk. Bioessays 2007, 29, 145–154. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic mechanisms and the mismatch concept of the developmental origins of health and disease. Pediatr. Res. 2007, 61, 5R–10R. [Google Scholar] [CrossRef]
- Miska, E.A.; Ferguson-Smith, A.C. Transgenerational inheritance: Models and mechanisms of non-DNA sequence-based inheritance. Science 2016, 354, 59–63. [Google Scholar] [CrossRef]
- Barker, D.J. In utero programming of chronic disease. Clin. Sci. 1998, 95, 115–128. [Google Scholar] [CrossRef]
- Gluckman, P.D.; Hanson, M.A.; Cooper, C.; Thornburg, K.L. Effect of in utero and early-life conditions on adult health and disease. N. Engl. J. Med. 2008, 359, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, L. Epigenetics and fetal metabolic programming: A call for integrated research on larger cohorts. Diabetes 2013, 62, 1026–1028. [Google Scholar] [CrossRef] [Green Version]
- Barres, R.; Zierath, J.R. The role of diet and exercise in the transgenerational epigenetic landscape of T2DM. Nat. Rev. Endocrinol. 2016, 12, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Dong, H.; Becker, A.S.; Dapito, D.H.; Modica, S.; Grandl, G.; Opitz, L.; Efthymiou, V.; Straub, L.G.; Sarker, G.; et al. Cold-induced epigenetic programming of the sperm enhances brown adipose tissue activity in the offspring. Nat. Med. 2018, 24, 1776. [Google Scholar] [CrossRef] [PubMed]
- Masuyama, H.; Hiramatsu, Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology 2012, 153, 2823–2830. [Google Scholar] [CrossRef] [Green Version]
- Masuyama, H.; Mitsui, T.; Nobumoto, E.; Hiramatsu, Y. The effects of high-fat diet exposure in utero on the obesogenic and diabetogenic traits through epigenetic changes in adiponectin and leptin gene expression for multiple generations in female mice. Endocrinology 2015, 156, 2482–2491. [Google Scholar] [CrossRef] [Green Version]
- Hua, X.; Xiong, J.W.; Zhang, Y.J.; Cao, X.Y.; Sun, P.; Wu, J.; Chen, L. Exposure of pregnant mice to triclosan causes hyperphagic obesity of offspring via the hypermethylation of proopiomelanocortin promoter. Arch. Toxicol. 2019, 93, 547–558. [Google Scholar] [CrossRef]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Müller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [Green Version]
- Hjort, L.; Martino, D.; Grunnet, L.G.; Naeem, H.; Maksimovic, J.; Olsson, A.H.; Zhang, C.; Ling, C.; Olsen, S.F.; Saffery, R.; et al. Gestational diabetes and maternal obesity are associated with epigenome-wide methylation changes in children. JCI Insight 2018, 3, e122572. [Google Scholar] [CrossRef]
- Caffrey, A.; Irwin, R.E.; McNulty, H.; Strain, J.J.; Lees-Murdock, D.J.; McNulty, B.A.; Ward, M.; Walsh, C.P.; Pentieva, K. Gene-specific DNA methylation in newborns in response to folic acid supplementation during the second and third trimesters of pregnancy: Epigenetic analysis from a randomized controlled trial. Am. J. Clin. Nutr. 2018, 107, 566–575. [Google Scholar] [CrossRef]
- Block, T.; El-Osta, A. Epigenetic programming, early life nutrition and the risk of metabolic disease. Atherosclerosis 2017, 266, 31–40. [Google Scholar] [CrossRef] [PubMed]
- Dolk, H.; McCullough, N.; Callaghan, S.; Casey, F.; Craig, B.; Given, J.; Loane, M.; Lagan, B.M.; Bunting, B.; Boyle, B.; et al. Risk factors for congenital heart disease: The Baby Hearts Study, a population-based case-control study. PLoS ONE 2020, 15, e0227908. [Google Scholar] [CrossRef] [Green Version]
- Ramos-Lopez, O.; Milagro, F.I.; Riezu-Boj, J.I.; Martinez, J.A. Epigenetic signatures underlying inflammation: An interplay of nutrition, physical activity, metabolic diseases, and environmental factors for personalized nutrition. Inflamm. Res. 2021, 70, 29–49. [Google Scholar] [CrossRef]
- Hosseini, B.; Berthon, B.S.; Saedisomeolia, A.; Starkey, M.R.; Collison, A.; Wark, P.A.B.; Wood, L.G. Effects of fruit and vegetable consumption on inflammatory biomarkers and immune cell populations: A systematic literature review and meta-analysis. Am. J. Clin. Nutr. 2018, 108, 136–155. [Google Scholar] [CrossRef] [Green Version]
- Palatnik, A.; Moosreiner, A.; Olivier-Van Stichelen, S. Consumption of non-nutritive sweeteners during pregnancy. Am. J. Obstet. Gynecol. 2020, 223, 211–218. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat. Med. 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Hollingsworth, J.W.; Maruoka, S.; Boon, K.; Garantziotis, S.; Li, Z.; Tomfohr, J.; Bailey, N.; Potts, E.N.; Whitehead, G.; Brass, D.M.; et al. In utero supplementation with methyl donors enhances allergic airway disease in mice. J. Clin. Investig. 2008, 118, 3462–3469. [Google Scholar] [CrossRef] [Green Version]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef] [Green Version]
- Kurotaki, D.; Kawase, W.; Sasaki, H.; Nakabayashi, J.; Nishiyama, A.; Morse, H.C., 3rd; Ozato, K.; Suzuki, Y.; Tamura, T. Epigenetic control of early dendritic cell lineage specification by the transcription factor IRF8 in mice. Blood 2019, 133, 1803–1813. [Google Scholar] [CrossRef] [Green Version]
- Spicuglia, S.; Zacarias-Cabeza, J.; Pekowska, A.; Ferrier, P. Epigenetic regulation of antigen receptor gene rearrangement. F1000 Biol. Rep. 2010, 2, 23. [Google Scholar] [CrossRef]
- Bergman, Y.; Cedar, H. A stepwise epigenetic process controls immunoglobulin allelic exclusion. Nat. Rev. Immunol. 2004, 4, 753–761. [Google Scholar] [CrossRef]
- Gutierrez, M.J.; Nino, G.; Hong, X.; Wang, X. Epigenomics and early life human humoral immunity: Novel paradigms and research pportunities. Front. Immunol. 2020, 11, 1766. [Google Scholar] [CrossRef] [PubMed]
- Crimi, E.; Benincasa, G.; Figueroa-Marrero, N.; Galdiero, M.; Napoli, C. Epigenetic susceptibility to severe respiratory viral infections and its therapeutic implications: A narrative review. Br. J. Anaesth. 2020, 125, 1002–1017. [Google Scholar] [CrossRef]
- Kwon, H.S.; Lim, H.W.; Wu, J.; Schnölzer, M.; Verdin, E.; Ott, M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J. Immunol. 2012, 188, 2712–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, G.; Song, X.; Fujimoto, S.; Piccirillo, C.A.; Nagai, Y.; Greene, M.I. Foxp3 Posttranslational modifications and Treg suppressive activity. Front. Immunol. 2019, 10, 2486. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Yang, C.; Pan, F. Posttranslational regulations of Foxp3 in Treg cells and their therapeutic applications. Front. Immunol. 2021, 12, 626172. [Google Scholar] [CrossRef]
- Beyer, M.; Thabet, Y.; Müller, R.U.; Sadlon, T.; Classen, S.; Lahl, K.; Basu, S.; Zhou, X.; Bailey-Bucktrout, S.L.; Krebs, W.; et al. Repression of the genome organizer SATB1 in regulatory T cells is required for suppressive function and inhibition of effector differentiation. Nat. Immunol. 2011, 12, 898–907. [Google Scholar] [CrossRef] [Green Version]
- Bettini, M.L.; Pan, F.; Bettini, M.; Finkelstein, D.; Rehg, J.E.; Floess, S.; Bell, B.D.; Ziegler, S.F.; Huehn, J.; Pardoll, D.M.; et al. Loss of epigenetic modification driven by the Foxp3 transcription factor leads to regulatory T cell insufficiency. Immunity 2012, 36, 717–730. [Google Scholar] [CrossRef] [Green Version]
- Tóth, D.M.; Ocskó, T.; Balog, A.; Markovics, A.; Mikecz, K.; Kovács, L.; Jolly, M.; Bukiej, A.A.; Ruthberg, A.D.; Vida, A.; et al. Amelioration of autoimmune arthritis in mice treated with the DNA methyltransferase inhibitor 5′-Azacytidine. Arthritis Rheumatol. 2019, 71, 1265–1275. [Google Scholar] [CrossRef] [Green Version]
- Gervin, K.; Vigeland, M.D.; Mattingsdal, M.; Hammerø, M.; Nygård, H.; Olsen, A.O.; Brandt, I.; Harris, J.R.; Undlien, D.E.; Lyle, R. DNA methylation and gene expression changes in monozygotic twins discordant for psoriasis: Identification of epigenetically dysregulated genes. PLoS Genet. 2012, 8, e1002454. [Google Scholar] [CrossRef]
- Rakyan, V.K.; Beyan, H.; Down, T.A.; Hawa, M.I.; Maslau, S.; Aden, D.; Daunay, A.; Busato, F.; Mein, C.A.; Manfras, B.; et al. Identification of type 1 diabetes-associated DNA methylation variable positions that precede disease diagnosis. PLoS Genet. 2011, 7, e1002300. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lei, L.; Massart, R.; Suderman, M.J.; Machnes, Z.; Elgbeili, G.; Laplante, D.P.; Szyf, M.; King, S. DNA methylation signatures triggered by prenatal maternal stress exposure to a natural disaster: Project Ice Storm. PLoS ONE 2014, 9, e107653. [Google Scholar] [CrossRef] [Green Version]
- Cao-Lei, L.; Dancause, K.N.; Elgbeili, G.; Massart, R.; Szyf, M.; Liu, A.; Laplante, D.P.; King, S. DNA methylation mediates the impact of exposure to prenatal maternal stress on BMI and central adiposity in children at age 13½ years: Project Ice Storm. Epigenetics 2015, 10, 749–761. [Google Scholar] [CrossRef]
- Sen, S.; Iyer, C.; Klebenov, D.; Histed, A.; Aviles, J.A.; Meydani, S.N. Obesity impairs cell-mediated immunity during the second trimester of pregnancy. Am. J. Obstet. Gynecol. 2013, 208, 139.e1–139.e8. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Chen, H.; Xu, Y.; An, C.; Zhong, L.; Wang, X.; Zhang, Y.; Chen, H.; Zhang, J.; Wang, Z. Fetal growth is associated with maternal fasting plasma glucose at first prenatal visit. PLoS ONE 2014, 9, e116352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sureshchandra, S.; Wilson, R.M.; Rais, M.; Marshall, N.E.; Purnell, J.Q.; Thornburg, K.L.; Messaoudi, I. Maternal Pregravid Obesity Remodels the DNA Methylation Landscape of Cord Blood Monocytes Disrupting Their Inflammatory Program. J. Immunol. 2017, 199, 2729–2744. [Google Scholar] [CrossRef] [Green Version]
- Wilson, R.M.; Marshall, N.E.; Jeske, D.R.; Purnell, J.Q.; Thornburg, K.; Messaoudi, I. Maternal obesity alters immune cell frequencies and responses in umbilical cord blood samples. Pediatr. Allergy Immunol. 2015, 26, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Nemoda, Z.; Massart, R.; Suderman, M.; Hallett, M.; Li, T.; Coote, M.; Cody, N.; Sun, Z.S.; Soares, C.N.; Turecki, G.; et al. Maternal depression is associated with DNA methylation changes in cord blood T lymphocytes and adult hippocampi. Transl. Psychiatry 2015, 5, e545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, S.; Gundavarapu, S.; Peña-Philippides, J.; Rir-Sima-ah, J.; Mishra, N.; Wilder, J.; Langley, R.J.; Smith, K.R.; Sopori, M.L. Prenatal secondhand cigarette smoke promotes Th2 polarization and impairs goblet cell differentiation and airway mucus formation. J. Immunol. 2011, 187, 4542–4552. [Google Scholar] [CrossRef]
- Savran, O.; Ulrik, C.S. Early life insults as determinants of chronic obstructive pulmonary disease in adult life. Int. J. Chron. Obstruct. Pulmon. Dis. 2018, 13, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drago, G.; Ruggieri, S.; Cuttitta, G.; La Grutta, S.; Ferrante, G.; Cibella, F. Determinants of Allergic Sensitization, Asthma and Lung Function: Results from a Cross-Sectional Study in Italian Schoolchildren. Int. J. Environ. Res. Public. Health 2020, 17, 5087. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.M.; Chiang, B.L.; Wang, L.C. Maternal Nutritional Status and Development of Atopic Dermatitis in Their Offspring. Clin. Rev. Allergy Immunol. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Venter, C.; Agostoni, C.; Arshad, S.H.; Ben-Abdallah, M.; Du Toit, G.; Fleischer, D.M.; Greenhawt, M.; Glueck, D.H.; Groetch, M.; Lunjani, N.; et al. Dietary factors during pregnancy and atopic outcomes in childhood: A systematic review from the European Academy of Allergy and Clinical Immunology. Pediatr. Allergy Immunol. 2020, 31, 889–912. [Google Scholar] [CrossRef]
- Ogawa, K.; Pak, K.; Yamamoto-Hanada, K.; Ishitsuka, K.; Sasaki, H.; Mezawa, H.; Saito-Abe, M.; Sato, M.; Yang, L.; Nishizato, M.; et al. Association between maternal vegetable intake during pregnancy and allergy in offspring: Japan Environment and Children’s Study. PLoS ONE 2021, 16, e0245782. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, N.; Alashkar Alhamwe, B.; Caraballo, L.; Ding, M.; Ferrante, A.; Garn, H.; Garssen, J.; Hii, C.S.; Irvine, J.; Llinás-Caballero, K.; et al. Perinatal and Early-Life Nutrition, Epigenetics, and Allergy. Nutrients 2021, 13, 724. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tu, C.; Yu, J.; Chen, M.; Tan, C.; Zheng, X.; Wang, Z.; Liang, Y.; Wang, K.; Wu, J.; et al. Maternal microbiome regulation prevents early allergic airway diseases in mouse offspring. Pediatr. Allergy Immunol. 2020, 31, 962–973. [Google Scholar] [CrossRef]
- Forsberg, A.; Abrahamsson, T.R.; Nilsson, L.; Ernerudh, J.; Duchén, K.; Jenmalm, M.C. Changes in peripheral immune populations during pregnancy and modulation by probiotics and ω-3 fatty acids. Sci. Rep. 2020, 10, 18723. [Google Scholar] [CrossRef]
- Fiuza, B.S.D.; Fonseca, H.F.; Meirelles, P.M.; Marques, C.R.; da Silva, T.M.; Figueiredo, C.A. Understanding Asthma and Allergies by the Lens of Biodiversity and Epigenetic Changes. Front. Immunol. 2021, 12, 623737. [Google Scholar] [CrossRef]
- Gunawardhana, L.P.; Baines, K.J.; Mattes, J.; Murphy, V.E.; Simpson, J.L.; Gibson, P.G. Differential DNA methylation profiles of infants exposed to maternal asthma during pregnancy. Pediatr. Pulmonol. 2014, 49, 852–862. [Google Scholar] [CrossRef]
- Wang, I.J.; Chen, S.L.; Lu, T.P.; Chuang, E.Y.; Chen, P.C. Prenatal smoke exposure, DNA methylation, and childhood atopic dermatitis. Clin. Exp. Allergy 2013, 43, 535–543. [Google Scholar] [CrossRef]
- Paplinska-Goryca, M.; Misiukiewicz-Stepien, P.; Proboszcz, M.; Nejman-Gryz, P.; Gorska, K.; Krenke, R. The Expressions of TSLP, IL-33, and IL-17A in Monocyte Derived Dendritic Cells from Asthma and COPD Patients are Related to Epithelial-Macrophage Interactions. Cells 2020, 9, 1944. [Google Scholar] [CrossRef] [PubMed]
- Paplińska-Goryca, M.; Nejman-Gryz, P.; Proboszcz, M.; Kwiecień, I.; Hermanowicz-Salamon, J.; Grabczak, E.M.; Krenke, R. Expression of TSLP and IL-33 receptors on sputum macrophages of asthma patients and healthy subjects. J. Asthma 2020, 57, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Berna, R.; Mitra, N.; Lou, C.; Wan, J.; Hoffstad, O.; Wubbenhorst, B.; Nathanson, K.L.; Margolis, D.J. TSLP and IL-7R Variants Are Associated with Persistent Atopic Dermatitis. J. Investig. Dermatol. 2021, 141, 446–450.e2. [Google Scholar] [CrossRef]
- O’Shea, K.M.; Aceves, S.S.; Dellon, E.S.; Gupta, S.K.; Spergel, J.M.; Furuta, G.T.; Rothenberg, M.E. Pathophysiology of Eosinophilic Esophagitis. Gastroenterology 2018, 154, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Jerram, S.T.; Leslie, R.D. The genetic architecture of type 1 diabetes. Genes 2017, 8, 209. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.S.; Hwang, J.S. Genetic aspects of type 1 diabetes. Ann. Pediatr. Endocrinol. Metab. 2019, 24, 143–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, J.C.; Clayton, D.G.; Concannon, P.; Akolkar, B.; Cooper, J.D.; Erlich, H.A.; Julier, C.; Morahan, G.; Nerup, J.; Nierras, C.; et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat. Genet. 2009, 41, 703–707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.; Deutsch, A.J.; Lenz, T.L.; Onengut-Gumuscu, S.; Han, B.; Chen, W.-M.; Howson, J.M.M.; Todd, J.A.; de Bakker, P.I.W.; Rich, S.S.; et al. Additive and interaction effects at three amino acid positions in HLA-DQ and HLA-DR molecules drive type 1 diabetes risk. Nat. Genet. 2015, 47, 898–905. [Google Scholar] [CrossRef] [Green Version]
- Van Lummel, M.; Van Veelen, P.A.; De Ru, A.H.; Janssen, G.M.; Pool, J.; Laban, S.; Joosten, A.M.; Nikolic, T.; Drijfhout, J.W.; Mearin, M.L. Dendritic Cells Guide Islet Autoimmunity through a Restricted and Uniquely Processed Peptidome Presented by High-Risk HLA-DR. J. Immunol. 2016, 196, 3253–3263. [Google Scholar] [CrossRef]
- Van Lummel, M.; van Veelen, P.A.; de Ru, A.H.; Pool, J.; Nikolic, T.; Laban, S.; Joosten, A.; Drijfhout, J.W.; Gómez-Touriño, I.; Arif, S.; et al. Discovery of a Selective Islet Peptidome Presented by the Highest-Risk HLA-DQ8trans Molecule. Diabetes 2016, 65, 732–741. [Google Scholar] [CrossRef] [Green Version]
- Nejentsev, S.; Howson, J.; Walker, N.M.; Szeszko, J.; Field, S.F.; Stevens, H.E.; Reynolds, P.; Hardy, M.; King, E.; Masters, J.; et al. Localization of type 1 diabetes susceptibility to the MHC class I genes HLA-B and HLA-A. Nature 2007, 450, 887–892. [Google Scholar] [CrossRef]
- Pugliese, A.; Zeller, M.; Fernandez, A., Jr.; Zalcberg, L.J.; Bartlett, R.J.; Ricordi, C.; Pietropaolo, M.; Eisenbarth, G.S.; Bennett, S.T.; Patel, D.D. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 1997, 15, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Vafiadis, P.; Bennett, S.T.; Todd, J.A.; Nadeau, J.; Grabs, R.; Goodyer, C.G.; Wickramasinghe, S.; Colle, E.; Polychronakos, C. Insulin expression in human thymus is modulated by INS VNTR alleles at the IDDM2 locus. Nat. Genet. 1997, 15, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Musumeci, L.; Alonso, A.; Rahmouni, S.; Nika, K.; Rostamkhani, M.; MacMurray, J.; Meloni, G.F.; Lucarelli, P.; Pellecchia, M.; et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat. Genet. 2004, 36, 337–338. [Google Scholar] [CrossRef] [PubMed]
- Vella, A.; Cooper, J.D.; Lowe, C.E.; Walker, N.; Nutland, S.; Widmer, B.; Jones, R.; Ring, S.M.; McArdle, W.; Pembrey, M.E.; et al. Localization of a type 1 diabetes locus in the IL2RA/CD25 region by use of tag single-nucleotide polymorphisms. Am. J. Hum. Genet. 2005, 76, 773–779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Muhlendahl, K.E.; Herkenhoff, H. Long-term course of neonatal diabetes. N. Engl. J. Med. 1995, 333, 704–708. [Google Scholar] [CrossRef] [PubMed]
- Shield, J.P.; Gardner, R.J.; Wadsworth, E.J.; Whiteford, M.L.; James, R.S.; Robinson, D.O.; Baum, J.D.; Temple, I.K. Aetiopathology and genetic basis of neonatal diabetes. Arch. Dis. Child. Fetal Neonatal 1997, 76, F39–F42. [Google Scholar] [CrossRef] [Green Version]
- Arima, T.; Kamikihara, T.; Hayashida, T.; Kato, K.; Inoue, T.; Shirayoshi, Y.; Oshimura, M.; Soejima, H.; Mukai, T.; Wake, N. ZAC, LIT1 (KCNQ1OT1) and p57KIP2 (CDKN1C) are in an imprinted gene network that may play a role in Beckwith-Wiedemann syndrome. Nucleic Acids Res. 2005, 33, 2650–2660. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Patch, A.M.; Mackay, D.J.; Edghill, E.L.; Gloyn, A.L.; Robinson, D.; Shield, J.P.; Temple, K.; Ellard, S.; Hattersley, A.T. Mutations in ATP-sensitive K+ channel genes cause transient neonatal diabetes and permanent diabetes in childhood or adulthood. Diabetes 2007, 56, 1930–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slingerland, A.S.; Shields, B.M.; Flanagan, S.E.; Bruining, G.J.; Noordam, K.; Gach, A.; Mlynarski, W.; Malecki, M.T.; Hattersley, A.T.; Ellard, S. Referral rates for diagnostic testing support an incidence of permanent neonatal diabetes in three European countries of at least 1 in 260,000 live births. Diabetologia 2009, 52, 1683–1685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackay, D.J.; Temple, I.K. Transient neonatal diabetes mellitus type 1. Am. J. Med. Genet. C. Semin. Med. Genet. 2010, 154C, 335–342. [Google Scholar] [CrossRef]
- Wallace, C.; Smyth, D.J.; Maisuria-Armer, M.; Walker, N.M.; Todd, J.A.; Clayton, D.G. The imprinted DLK1-MEG3 gene region on chromosome 14q32.2 alters susceptibility to type 1 diabetes. Nat. Genet. 2010, 42, 68–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, A.; Steinthorsdottir, V.; Masson, G.; Thorleifsson, G.; Sulem, P.; Besenbacher, S.; Jonasdottir, A.; Sigurdsson, A.; Kristinsson, K.T.; Jonasdottir, A.; et al. Parental origin of sequence variants associated with complex diseases. Nature 2009, 462, 868–874. [Google Scholar] [CrossRef] [Green Version]
- Tuomilehto, J. The emerging global epidemic of type 1 diabetes. Curr. Diabetes Rep. 2013, 13, 795–804. [Google Scholar] [CrossRef]
- Kyvik, K.O.; Green, A.; Beck-Nielsen, H. Concordance rates of insulin dependent diabetes mellitus: A population based study of young Danish twins. BMJ 1995, 311, 913–917. [Google Scholar] [CrossRef] [Green Version]
- Fava, D.; Gardner, S.; Pyke, D.; Leslie, R.D. Evidence that the age at diagnosis of IDDM is genetically determined. Diabetes Care 1998, 21, 925–929. [Google Scholar] [CrossRef]
- Metcalfe, K.A.; Hitman, G.A.; Rowe, R.E.; Hawa, M.; Huang, X.; Stewart, T.; Leslie, R.D. Concordance for type 1 diabetes in identical twins is affected by insulin genotype. Diabetes Care 2001, 24, 838–842. [Google Scholar] [CrossRef] [Green Version]
- Redondo, M.J.; Yu, L.; Hawa, M.; Mackenzie, T.; Pyke, D.A.; Eisenbarth, G.S.; Leslie, R.D. Heterogeneity of type I diabetes: Analysis of monozygotic twins in Great Britain and the United States. Diabetologia 2001, 44, 354–362. [Google Scholar] [CrossRef]
- Hyttinen, V.; Kaprio, J.; Kinnunen, L.; Koskenvuo, M.; Tuomilehto, J. Genetic liability of type 1 diabetes and the onset age among 22,650 young Finnish twin pairs: A nationwide follow-up study. Diabetes 2003, 52, 1052–1055. [Google Scholar] [CrossRef] [Green Version]
- Triolo, T.M.; Fouts, A.; Pyle, L.; Yu, L.; Gottlieb, P.A.; Steck, A.K.; Type 1 Diabetes TrialNet Study Group. Identical and Nonidentical Twins: Risk and factors involved in development of islet autoimmunity and type 1 diabetes. Diabetes Care 2019, 42, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Blunk, I.; Thomsen, H.; Reinsch, N.; Mayer, M.; Försti, A.; Sundquist, J.; Sundquist, K.; Hemminki, K. Genomic imprinting analyses identify maternal effects as a cause of phenotypic variability in type 1 diabetes and rheumatoid arthritis. Sci. Rep. 2020, 10, 11562. [Google Scholar] [CrossRef]
- Söderström, U.; Aman, J.; Hjern, A. Being born in Sweden increases the risk for type 1 diabetes-a study of migration of children to Sweden as a natural experiment. Acta Paediatr. 2012, 101, 73–77. [Google Scholar] [CrossRef]
- Malmqvist, E.; Larsson, H.E.; Jönsson, I.; Rignell-Hydbom, A.; Ivarsson, S.A.; Tinnerberg, H.; Stroh, E.; Rittner, R.; Jakobsson, K.; Swietlicki, E.; et al. Maternal exposure to air pollution and type 1 diabetes-Accounting for genetic factors. Environ. Res. 2015, 140, 268–274. [Google Scholar] [CrossRef] [Green Version]
- Vasudevan, D.; Hickok, J.R.; Bovee, R.C.; Pham, V.; Mantell, L.L.; Bahroos, N.; Kanabar, P.; Cao, X.J.; Maienschein-Cline, M.; Garcia, B.; et al. Nitric oxide regulates gene expression in cancers by controlling histone posttranslational modifications. Cancer Res. 2015, 5, 1582. [Google Scholar] [CrossRef] [Green Version]
- Fry, R.C.; Rager, J.E.; Bauer, R.; Sebastian, E.; Peden, D.B.; Jaspers, I.; Alexis, N.E. Air toxics and epigenetic effects: Ozone altered microRNAs in the sputum of human subjects. Am. J. Physiol. Lung. Cell. Mol. Physiol. 2014, 306, L1129–L1137. [Google Scholar] [CrossRef] [Green Version]
- Mattsson, K.; Jönsson, I.; Malmqvist, E.; Larsson, H.E.; Rylander, L. Maternal smoking during pregnancy and offspring type 1 diabetes mellitus risk: Accounting for HLA haplotype. Eur J. Epidemiol. 2015, 30, 231–238. [Google Scholar] [CrossRef]
- Gundersen, E. Is diabetes of infectious origin? J. Infect. Dis. 1927, 41, 197–202. [Google Scholar] [CrossRef]
- Sarmiento, L.; Cubas-Dueñas, I.; Cabrera-Rode, E. Evidence of association between type 1 diabetes and exposure to enterovirus in Cuban children and adolescents. MEDICC Rev. 2013, 15, 29–32. [Google Scholar] [CrossRef]
- Geravandi, S.; Liu, H.; Maedler, K. Enteroviruses and T1D: Is It the Virus, the Genes or Both which Cause T1D. Microorganisms 2020, 8, 1017. [Google Scholar] [CrossRef]
- Dahlquist, G.; Frisk, G.; Ivarsson, S.A.; Svanberg, L.; Forsgren, M.; Diderholm, H. Indications that maternal coxsackie B virus infection during pregnancy is a risk factor for childhood- onset IDDM. Diabetologia 1995, 38, 1371–1373. [Google Scholar] [CrossRef]
- Dahlquist, G.G.; Ivarsson, S.; Lingberg, B.; Forsgren, M. Maternal enteroviral infection in pregnancy is a risk factor for childhood IDDM-A population based case control study. Diabetes 1995, 44, 408–413. [Google Scholar] [CrossRef]
- Dalhquist, G.; Kalen, B. Time-space clustering of time at birth in childhood onset diabetes. Diabetes Care 1996, 19, 328–332. [Google Scholar] [CrossRef]
- Dahlquist, G. Viruses and other perinatal exposure as initiating events for β-cell destruction. Ann. Med. 1997, 29, 413–417. [Google Scholar] [CrossRef] [PubMed]
- Dahlquist, G.G.; Bowman, J.E.; Juto, P. Enteroviral RNA and IGM antibodies in early pregnancy is a risk factor for childhood onset IDDM. Diabetes Care 1999, 22, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Viskari, H.R.; Roivainen, M.; Reunanen, A.; Pitkäniemi, J.; Sadeharju, K.; Koskela, P.; Hovi, T.; Leinikki, P.; Vilja, P.; Tuomilehto, J.; et al. Maternal first-trimester enterovirus infection and future risk of type 1 diabetes in the exposed fetus. Diabetes 2002, 51, 2568–2571. [Google Scholar] [CrossRef] [Green Version]
- Dahlquist, G.G.; Forsberg, J.; Hagenfeldt, L.; Boman, J.; Juto, P. Increased prevalence of enteroviral RNA in blood spots from newborn children who later developed type 1 diabetes: A population-based case-control study. Diabetes Care 2004, 27, 285–286. [Google Scholar] [CrossRef] [Green Version]
- Elfving, M.; Svensson, J.; Oikarinen, S.; Jonsson, B.; Olofsson, P.; Sundkvist, G.; Lindberg, B.; Lernmark, A.; Hyöty, H.; Ivarsson, S.A. Maternal enterovirus infection during pregnancy as a risk factor in offspring diagnosed with type 1diabetes between 15 and 30 years of age. Exp. Diabetes Res. 2008, 2008, 271958. [Google Scholar] [CrossRef] [Green Version]
- Rešić Lindehammer, S.; Honkanen, H.; Nix, W.A.; Oikarinen, M.; Lynch, K.F.; Jönsson, I.; Marsal, K.; Oberste, S.; Hyöty, H.; Lernmark, Å. Seroconversion to islet autoantibodies after enterovirus infection in early pregnancy. Viral Immunol. 2012, 25, 254–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viskari, H.; Knip, M.; Tauriainen, S.; Huhtala, H.; Veijola, R.; Ilonen, J.; Simell, O.; Surcel, H.M.; Hyöty, H. Maternal enterovirus infection as a risk factor for type 1 diabetes in the exposed offspring. Diabetes Care 2012, 35, 1328–1332. [Google Scholar] [CrossRef] [Green Version]
- Penno, M.; Couper, J.J.; Craig, M.E.; Colman, P.G.; Rawlinson, W.D.; Cotterill, A.M.; Jones, T.W.; Harrison, L.C.; ENDIA Study Group. Environmental determinants of islet autoimmunity (ENDIA): A pregnancy to early life cohort study in children at-risk of type 1 diabetes. BMC Pediatr. 2013, 13, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, D.W.; Kim, K.; Rawlinson, W.D.; Craig, M.E. Maternal virus infections in pregnancy and type 1 diabetes in their offspring: Systematic review and meta-analysis of observational studies. Rev. Med. Virol. 2018, 28, e1974. [Google Scholar] [CrossRef] [PubMed]
- Wook Kim, K.; Allen, D.W.; Briese, T.; Couper, J.J.; Barry, S.C.; Colman, P.G.; Cotterill, A.M.; Davis, E.A.; Giles, L.C.; Harrison, L.C.; et al. Distinct gut virome profile of pregnant women with type 1 diabetes in the ENDIA study. Open Forum Infect. Dis. 2019, 6, ofz025. [Google Scholar] [CrossRef] [PubMed]
- Craig, M.E.; Kim, K.W.; Isaacs, S.R.; Penno, M.A.; Hamilton-Williams, E.E.; Couper, J.J.; Rawlinson, W.D. Early-life factors contributing to type 1 diabetes. Diabetologia 2019, 62, 1823–1834. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.W.; Ho, A.; Alshabee-Akil, A.; Hardikar, A.A.; Kay, T.W.; Rawlinson, W.D.; Craig, M.E. Coxsackievirus B5 Infection Induces Dysregulation of microRNAs Predicted to Target Known Type 1 Diabetes Risk Genes in Human Pancreatic Islets. Diabetes 2016, 65, 996–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alidjinou, E.K.; Engelmann, I.; Bossu, J.; Villenet, C.; Figeac, M.; Romond, M.B.; Sané, F.; Hober, D. Persistence of Coxsackievirus B4 in pancreatic ductal-like cells results in cellular and viral changes. Virulence 2017, 8, 1229–1244. [Google Scholar] [CrossRef]
- Engelmann, I.; Alidjinou, E.K.; Bertin, A.; Bossu, J.; Villenet, C.; Figeac, M.; Sane, F.; Hober, D. Persistent coxsackievirus B4 infection induces microRNA dysregulation in human pancreatic cells. Cell. Mol. Life Sci. 2017, 74, 3851–3861. [Google Scholar] [CrossRef] [PubMed]
- McErlean, P.; Favoreto, S., Jr.; Costa, F.F.; Shen, J.; Quraishi, J.; Biyasheva, A.; Cooper, J.J.; Scholtens, D.M.; Vanin, E.F.; De Bonaldo, M.F.; et al. Human rhinovirus infection causes different DNA methylation changes in nasal epithelial cells from healthy and asthmatic subjects. BMC Med. Genom. 2014, 7, 37. [Google Scholar] [CrossRef] [Green Version]
- Bell, C.G.; Teschendorff, A.E.; Rakyan, V.K.; Maxwell, A.P.; Beck, S.; Savage, D.A. Genome-wide DNA methylation analysis for diabetic nephropathy in type 1 diabetes mellitus. BMC Med. Genom. 2010, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Fradin, D.; Le, F.S.; Mille, C.; Naoui, N.; Groves, C.; Zelenika, D.; McCarthy, M.I.; Lathrop, M.; Bougnères, P. Association of the CpG methylation pattern of the proximal insulin gene promoter with type 1 diabetes. PLoS ONE 2012, 7, e36278. [Google Scholar] [CrossRef]
- Belot, M.P.; Fradin, D.; Mai, N.; Le Fur, S.; Zélénika, D.; Kerr-Conte, J.; Pattou, F.; Lucas, B.; Bougnères, P. CpG methylation changes within the IL2RA promoter in type 1 diabetes of childhood onset. PLoS ONE 2013, 8, e68093. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.; Falhammar, H.; Gu, H.F.; Brismar, K. Epigenetic analyses of the insulin-like growth factor binding protein 1 gene in type 1 diabetes and diabetic nephropathy. Clin. Epigenet. 2014, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Agardh, E.; Lundstig, A.; Perfilyev, A.; Volkov, P.; Freiburghaus, T.; Lindholm, E.; Rönn, T.; Agardh, C.D.; Ling, C. Genome-wide analysis of DNA methylation in subjects with type 1 diabetes identifies epigenetic modifications associated with proliferative diabetic retinopathy. BMC Med. 2015, 13, 182. [Google Scholar] [CrossRef] [Green Version]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7, 13555. [Google Scholar] [CrossRef]
- Sklenarova, J.; Petruzelkova, L.; Kolouskova, S.; Lebl, J.; Sumnik, Z.; Cinek, O. Glucokinase gene may be a more suitable target than the insulin gene for detection of β cell death. Endocrinology 2017, 158, 2058–2065. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Richardson, T.G.; McArdle, W.L.; Relton, C.L.; Gillespie, K.M.; Suderman, M.; Hemani, G. Identification of loci where DNA methylation potentially mediates genetic risk of type 1 diabetes. J. Autoimmun. 2018, 93, 66–75. [Google Scholar] [CrossRef] [Green Version]
- Simmons, K.M.; Fouts, A.; Pyle, L.; Clark, P.; Dong, F.; Yu, L.; Usmani-Brown, S.; Gottlieb, P.; Herold, K.C.; Steck, A.K. Type 1 diabetes TrialNet Study Group. Unmethylated insulin as an adjunctive marker of β cell death and progression to type 1 diabetes in participants at risk for diabetes. Int. J. Mol. Sci. 2019, 20, 3857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carry, P.M.; Vanderlinden, L.A.; Johnson, R.K.; Dong, F.; Steck, A.K.; Frohnert, B.I.; Rewers, M.; Yang, I.V.; Kechris, K.; Norris, J.M. DNA methylation near the INS gene is associated with INS genetic variation (rs689) and type 1 diabetes in the Diabetes Autoimmunity Study in the Young. Pediatr. Diabetes 2020, 21, 597–605. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Stefan-Lifshitz, M.; Tomer, Y. Genetic and environmental factors regulate the type 1 diabetes gene CTSH via differential DNA methylation. J. Biol. Chem. 2021, 13, 100774. [Google Scholar] [CrossRef] [PubMed]
- Ludvigsson, J.; Faresjö, M.; Hjorth, M.; Axelsson, S.; Chéramy, M.; Pihl, M.; Vaarala, O.; Forsander, G.; Ivarsson, S.; Johansson, C.; et al. GAD treatment and insulin secretion in recent-onset type 1 diabetes. N. Engl. J. Med. 2008, 359, 1909–1920. [Google Scholar] [CrossRef] [Green Version]
- Cepek, P.; Zajacova, M.; Kotrbova-Kozak, A.; Silhova, E.; Cerna, M. DNA methylation and mRNA expression of HLA-DQA1 alleles in type 1 diabetes mellitus. Immunology 2016, 148, 150–159. [Google Scholar] [CrossRef] [Green Version]
- Van Kerckhove, C. Lupus erythematosus in childhood: Effect of maternal factors beyond neonatal disease? Clin. Rheumatol. 1999, 9, 168–170. [Google Scholar] [CrossRef]
- Erden, A.; Fanouriakis, A.; Kiliç, L.; Sari, A.; Armağan, B.; Bilgin, E.; Şener, Y.Z.; Hymabaccus, B.; Gürler, F.; Ceylan, S.; et al. Geoepidemiology and clinical characteristics of neonatal lupus erythematosus: A systematic literature review of individual patients’ data. Turk. J. Med. Sci. 2020, 50, 281–290. [Google Scholar] [CrossRef]
- Joshi, K.; Zacharin, M. Hyperthyroidism in an infant of a mother with autoimmune hypothyroidism with positive TSH receptor antibodies. J. Pediatr. Endocrinol. Metab. 2018, 31, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Delay, F.; Dochez, V.; Biquard, F.; Cheve, M.T.; Gillard, P.; Arthuis, C.J.; Winer, N. Management of fetal goiters: 6-year retrospective observational study in three prenatal diagnosis and treatment centers of the Pays De Loire Perinatal Network. J. Matern. Fetal Neonatal Med. 2020, 33, 2561–2569. [Google Scholar] [CrossRef]
- Greeley, S.A.; Katsumata, M.; Yu, L.; Eisenbarth, G.S.; Moore, D.J.; Goodarzi, H.; Barker, C.F.; Naji, A.; Noorchashm, H. Elimination of maternally transmitted autoantibodies prevents diabetes in nonobese diabetic mice. Nat. Med. 2002, 8, 399–402. [Google Scholar] [CrossRef]
- Silva, D.G.; Daley, S.R.; Hogan, J.; Lee, S.K.; The, C.E.; Hu, D.Y.; Lam, K.P.; Goodnow, C.C.; Vinuesa, C.G. Anti-islet autoantibodies trigger autoimmune diabetes in the presence of an increased frequency of islet-reactive CD4 T cells. Diabetes 2011, 60, 2102–2111. [Google Scholar] [CrossRef] [Green Version]
- Koczwara, K.; Ziegler, A.G.; Bonifacio, E. Maternal immunity to insulin does not affect diabetes risk in progeny of non-obese diabetic mice. Clin. Exp. Immunol. 2004, 136, 56–59. [Google Scholar] [CrossRef]
- Lindberg, B.; Ivarsson, S.A.; Landin-Olsson, M.; Sundkvist, G.; Svanberg, L.; Lernmark, A. Islet autoantibodies in cord blood from children who developed type I (insulin-dependent) diabetes mellitus before 15 years of age. Diabetologia 1999, 42, 181–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundgren, M.; Lynch, K.; Larsson, C.; Elding Larsson, H.; Diabetes Prediction in Skåne study group. Cord blood insulinoma-associated protein 2 autoantibodies are associated with increased risk of type 1 diabetes in the population-based Diabetes Prediction in Skåne study. Diabetologia 2015, 58, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koczwara, K.; Bonifacio, E.; Ziegler, A.G. Transmission of maternal islet antibodies and risk of autoimmune diabetes in offspring of mothers with type 1 diabetes. Diabetes 2004, 53, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Warram, J.H.; Krolewski, A.S.; Gottlieb, M.S.; Kahn, C.R. Differences in risk of insulin-dependent diabetes in offspring of diabetic mothers and diabetic fathers. N. Engl. J. Med. 1984, 311, 149–152. [Google Scholar] [CrossRef]
- Warram, J.H.; Krolewski, A.S.; Kahn, C.R. Determinants of IDDM and perinatal mortality in children of diabetic mothers. Diabetes 1988, 37, 1328–1334. [Google Scholar] [CrossRef]
- Warram, J.H.; Martin, B.C.; Krolewski, A.S. Risk of IDDM in children of diabetic mothers decreases with increasing maternal age at pregnancy. Diabetes 1991, 40, 1679–1684. [Google Scholar] [CrossRef] [PubMed]
- Dahlquist, G.; Blom, L.; Lönnberg, G. The Swedish Childhood Diabetes Study-a multivariate analysis of risk determinants for diabetes in different age groups. Diabetologia 1991, 34, 757–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pociot, F.; Nørgaard, K.; Hobolth, N.; Andersen, O.; Nerup, J.; Danish Study Group of Diabetes in Childhood. A nationwide population-based study of the familial aggregation of type 1 (insulin-dependent) diabetes mellitus in Denmark. Diabetologia 1993, 36, 870–875. [Google Scholar] [CrossRef] [Green Version]
- Harjutsalo, V.; Reunanen, A.; Tuomilehto, J. Differential transmission of type 1 diabetes from diabetic fathers and mothers to their offspring. Diabetes 2006, 55, 1517–1524. [Google Scholar] [CrossRef] [Green Version]
- Holm, B.C.; Svensson, J.; Akesson, C.; Arvastsson, J.; Ljungberg, J.; Lynch, K.; Ivarsson, S.A.; Lernmark, A.; Cilio, C.M.; Diabetes Prediction Study in Skåne (DiPiS). Evidence for immunological priming and increased frequency of CD4+ CD25+ cord blood T cells in children born to mothers with type 1 diabetes. Clin. Exp. Immunol. 2006, 146, 493–502. [Google Scholar] [CrossRef]
- Lentini, A.; Nestor, C.E. Mapping DNA methylation in mammals: The state of the Art. Methods Mol. Biol. 2021, 2198, 37–50. [Google Scholar] [CrossRef]


{kind=link}
| Reference/Year | Method | Sample | Results |
|---|---|---|---|
| [139]/2010 | Genome-wide DNA methylation | Whole blood | Association of 19 CpG sites with risk of diabetic nephropathy |
| [62]/2011 | Epigenome-wide association study (EWAS) | Monocytes | Presence of T1D-specific methylation variable positions in the T1D-affected co-twins |
| [140]/2012 | Methylation of specific genes | Whole blood | Association of CpG methylation at the INS locus with T1D |
| [141]/2013 | Methylation of specific genes | Peripheral blood | Effect of IL2RA risk alleles on T1D may be partially mediated through CpG methylation change |
| [142]/2014 | Methylation of specific genes | Peripheral blood | Decreased IGFBP1 DNA methylation levels are associated with T1D |
| [143]/2015 | Genome-wide DNA methylation | Whole blood | Subjects with T1D and proliferative diabetic retinopathy exhibit altered DNA methylation patterns in blood |
| [144]/2016 | Epigenome-wide association study | T cells B cells Monocytes | T1D-associated differentially variable CpG positions are located in genes involved in immune cell metabolism |
| [145]/2017 | Methylation of specific genes | Tissue, pancreatic islets, whole blood | Unmethylated glucokinase gene is more islet-specific than unmethylated INS DNA |
| [146]/2018 | Genome-wide DNA methylation | Whole blood | Methylation mediates T1D risk at five non-HLA loci mainly by influencing local gene expression. |
| [147]/2019 | Methylation of specific genes | Serum | A higher unmethylated INS ratio is associated with IAA levels at the time of T1D diagnosis |
| [148]/2020 | Methylation quantitative trait loci (mQTL) analyses | Peripheral blood | Identification of 10 single nucleotide polymorphism probe pairs significantly related to methylation levels prior to the development of T1D |
| [149]/2021 | Methylation of specific genes | Pancreatic islets | Pro-inflammatorycytokines and T1D genetic risk variants regulate CTSH transcription by differential DNA methylation |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barchetta, I.; Arvastsson, J.; Sarmiento, L.; Cilio, C.M. Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes. Genes 2021, 12, 887. https://doi.org/10.3390/genes12060887
Barchetta I, Arvastsson J, Sarmiento L, Cilio CM. Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes. Genes. 2021; 12(6):887. https://doi.org/10.3390/genes12060887
Chicago/Turabian StyleBarchetta, Ilaria, Jeanette Arvastsson, Luis Sarmiento, and Corrado M. Cilio. 2021. "Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes" Genes 12, no. 6: 887. https://doi.org/10.3390/genes12060887
APA StyleBarchetta, I., Arvastsson, J., Sarmiento, L., & Cilio, C. M. (2021). Epigenetic Changes Induced by Maternal Factors during Fetal Life: Implication for Type 1 Diabetes. Genes, 12(6), 887. https://doi.org/10.3390/genes12060887