
Analysis of Genetic Variants Associated with Levels of Immune Modulating Proteins for Impact on Alzheimer’s Disease Risk Reveal a Potential Role for SIGLEC14

 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of gDNA, RNA, and cDNA from Human Tissue
2.2. Genotyping and Copy Number Variant Assays
2.3. Gene Expression by qPCR
2.4. WGS Data Analysis
2.5. Statistical Analyses
3. Results
3.1. ITIM/ITAM pQTLs Are Overrepresented in AD GWAS Results
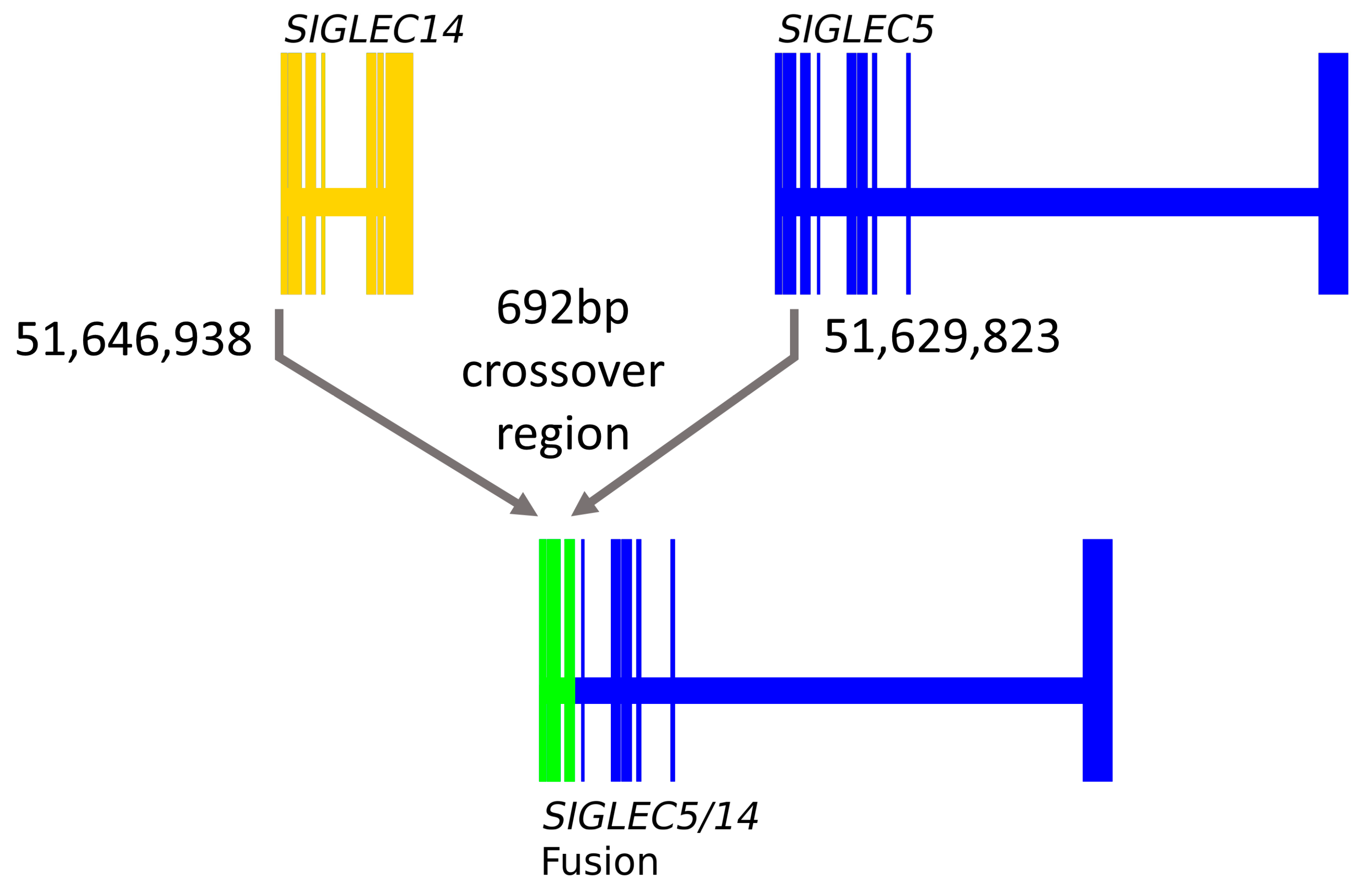
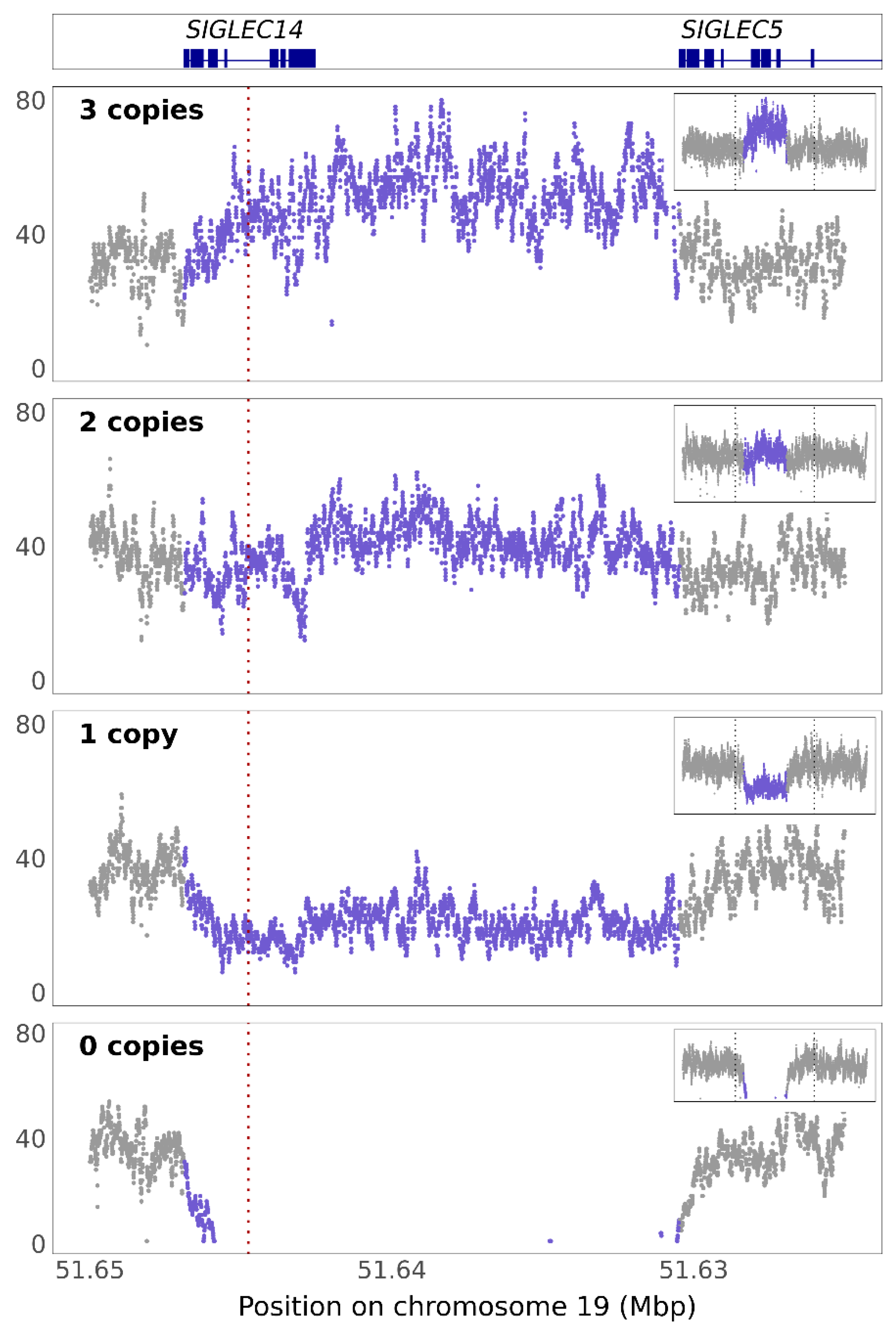
3.2. SIGLEC14 pQTL Is a Proxy for the Deletion Polymorphism
3.3. SIGLEC14 CNV Is Not Fully Captured by rs1106476
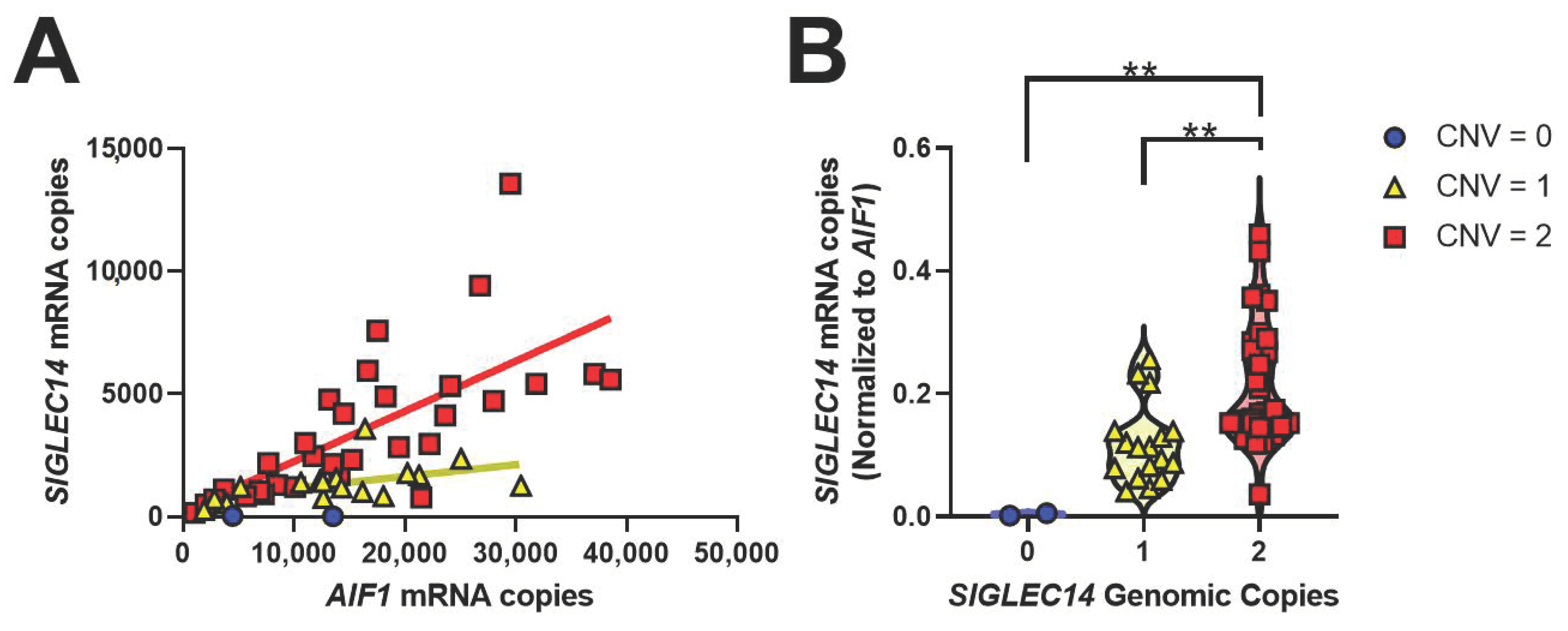
3.4. SIGLEC14 Is Expressed in Human Brain, and CNV Correlates with Gene Expression
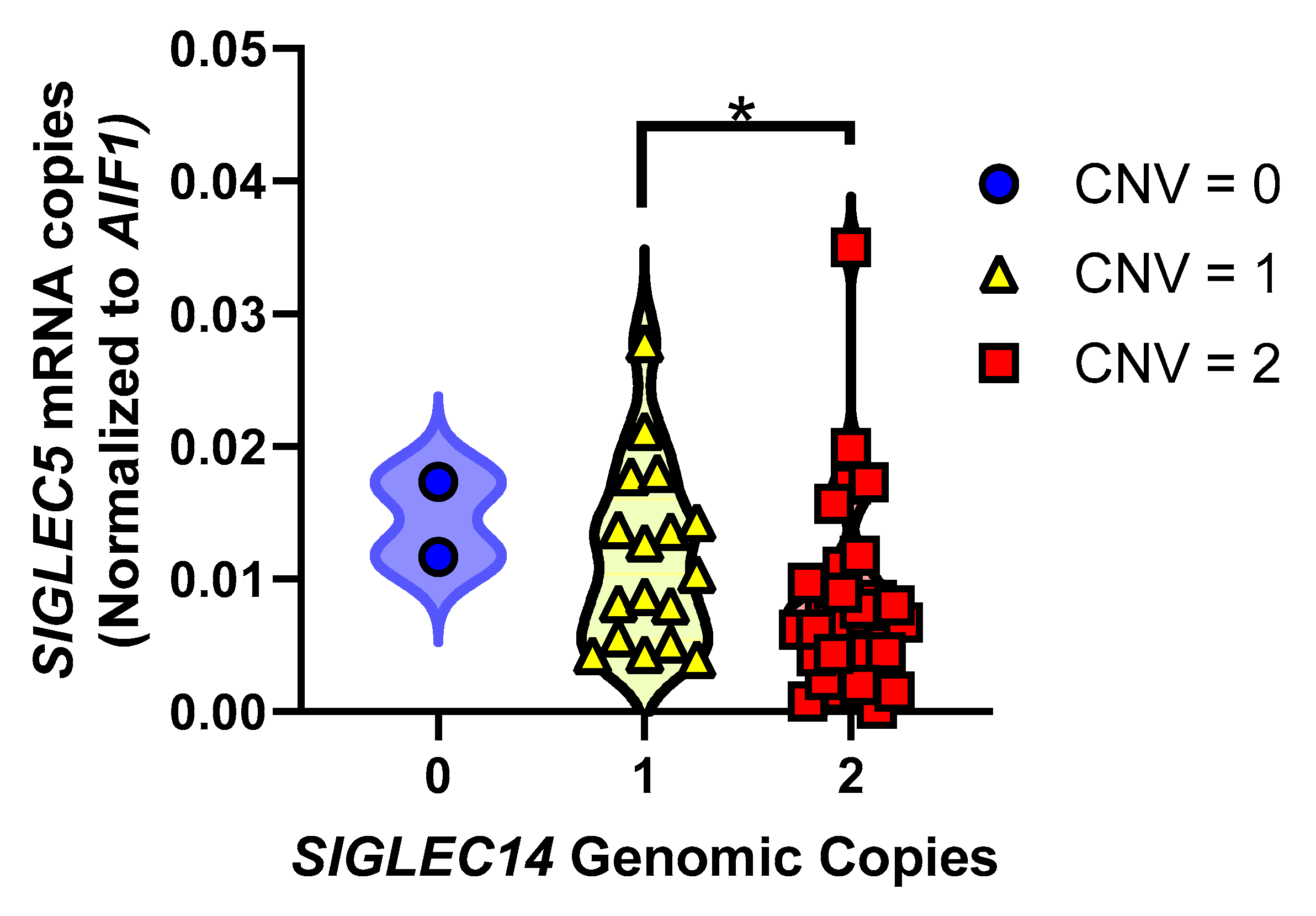
3.5. SIGLEC14 Deletion Leads to Increased SIGLEC5 Expression
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hagg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Kunkle, B.W.; Grenier-Boley, B.; Sims, R.; Bis, J.C.; Damotte, V.; Naj, A.C.; Boland, A.; Vronskaya, M.; van der Lee, S.J.; Amlie-Wolf, A.; et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat. Genet. 2019, 51, 414–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambert, J.C.; Ibrahim-Verbaas, C.A.; Harold, D.; Naj, A.C.; Sims, R.; Bellenguez, C.; DeStafano, A.L.; Bis, J.C.; Beecham, G.W.; Grenier-Boley, B.; et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat. Genet. 2013, 45, 1452–1458. [Google Scholar] [CrossRef] [Green Version]
- Naj, A.C.; Jun, G.; Beecham, G.W.; Wang, L.S.; Vardarajan, B.N.; Buros, J.; Gallins, P.J.; Buxbaum, J.D.; Jarvik, G.P.; Crane, P.K.; et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat. Genet. 2011, 43, 436–441. [Google Scholar] [CrossRef] [Green Version]
- Novikova, G.; Kapoor, M.; Tcw, J.; Abud, E.M.; Efthymiou, A.G.; Chen, S.X.; Cheng, H.; Fullard, J.F.; Bendl, J.; Liu, Y.; et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 2021, 12, 1–14. [Google Scholar] [CrossRef]
- Jones, L.; Holmans, P.A.; Hamshere, M.L.; Harold, D.; Moskvina, V.; Ivanov, D.; Pocklington, A.; Abraham, R.; Hollingworth, P.; Sims, R.; et al. Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS ONE 2010, 5, e13950. [Google Scholar] [CrossRef]
- Karch, C.M.; Goate, A.M. Alzheimer’s disease risk genes and mechanisms of disease pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhattacherjee, A.; Jung, S.J.; Ho, M.; Eskandari-Sedighi, G.; St. Laurent, C.D.; McCord, K.A.; Bains, A.; Gaurav, S.; Sarkar, S.S.; Plemel, J.; et al. The CD33 short isoform is a gain-of-function variant that enhances Aβ1-42 phagocytosis in microglia. Mol. Neurodegener. 2021, 16, 1–22. [Google Scholar] [CrossRef]
- Bhattacherjee, A.; Rodrigues, E.; Jung, J.; Luzentales-Simpson, M.; Enterina, J.R.; Galleguillos, D.; St. Laurent, C.D.; Nakhaei-Nejad, M.; Fuchsberger, F.F.; Streith, L.; et al. Repression of phagocytosis by human CD33 is not conserved with mouse CD33. Commun. Biol. 2019, 2, 450. [Google Scholar] [CrossRef] [Green Version]
- Chan, G.; White, C.C.; Winn, P.A.; Cimpean, M.; Replogle, J.M.; Glick, L.R.; Cuerdon, N.E.; Ryan, K.J.; Johnson, K.A.; Schneider, J.A.; et al. CD33 modulates TREM2: Convergence of Alzheimer loci. Nat. Neurosci. 2015, 18, 1556–1558. [Google Scholar] [CrossRef] [Green Version]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s Disease Risk Gene CD33 Inhibits Microglial Uptake of Amyloid Beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, M.; Chiles, I.I.I.J.; Xi, H.S.; Medway, C.; Simpson, J.; Potluri, S.; Howard, D.; Liang, Y.; Paumi, C.M.; Mukherjee, S.; et al. Genetics of CD33 in Alzheimer’s disease and acute myeloid leukemia. Hum. Mol. Genet. 2015, 24, 3557–3570. [Google Scholar] [CrossRef] [Green Version]
- Malik, M.; Simpson, J.F.; Parikh, I.; Wilfred, B.R.; Fardo, D.W.; Nelson, P.T.; Estus, S. CD33 Alzheimer’s Risk-Altering Polymorphism, CD33 Expression, and Exon 2 Splicing. J. Neurosci. 2013, 33, 13320–13325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, T.; Ryan, K.J.; Replogle, J.M.; Chibnik, L.B.; Rosenkrantz, L.; Tang, A.; Rothamel, K.; Stranger, B.E.; Bennett, D.A.; Evans, D.A.; et al. CD33: Increased inclusion of exon 2 implicates the Ig V-set domain in Alzheimer’s disease susceptibility. Hum. Mol. Genet. 2014, 23, 2729–2736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siddiqui, S.S.; Springer, S.A.; Verhagen, A.; Sundaramurthy, V.; Alisson-Silva, F.; Jiang, W.; Ghosh, P.; Varki, A. The Alzheimer’s disease-protective CD33 splice variant mediates adaptive loss of function via diversion to an intracellular pool. J. Biol. Chem. 2017, 292, 15312–15320. [Google Scholar] [CrossRef] [Green Version]
- McQuade, A.; Kang, Y.J.; Hasselmann, J.; Jairaman, A.; Sotelo, A.; Coburn, M.; Shabestari, S.K.; Chadarevian, J.P.; Fote, G.; Tu, C.H.; et al. Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer’s disease. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Estus, S.; Shaw, B.C.; Devanney, N.; Katsumata, Y.; Press, E.E.; Fardo, D.W. Evaluation of CD33 as a genetic risk factor for Alzheimer’s disease. Acta Neuropathol. 2019, 138, 187–199. [Google Scholar] [CrossRef]
- Griciuc, A.; Tanzi, R.E. The role of innate immune genes in Alzheimer’s disease. Curr. Opin. Neurol. 2021, 34, 228–236. [Google Scholar] [CrossRef]
- Efthymiou, A.G.; Goate, A.M. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol. Neurodegener. 2017, 12, 43. [Google Scholar] [CrossRef]
- Gandy, S.; Heppner, F.L. Microglia as dynamic and essential components of the amyloid hypothesis. Neuron 2013, 78, 575–577. [Google Scholar] [CrossRef] [Green Version]
- Allen, M.; Zou, F.; Chai, H.S.; Younkin, C.S.; Crook, J.; Pankratz, V.S.; Carrasquillo, M.M.; Rowley, C.N.; Nair, A.A.; Middha, S.; et al. Novel late-onset Alzheimer disease loci variants associate with brain gene expression. Neurology 2012, 79, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.B.; Maranville, J.C.; Peters, J.E.; Stacey, D.; Staley, J.R.; Blackshaw, J.; Burgess, S.; Jiang, T.; Paige, E.; Surendran, P.; et al. Genomic atlas of the human plasma proteome. Nature 2018, 558, 73–79. [Google Scholar] [CrossRef]
- Yamanaka, M.; Kato, Y.; Angata, T.; Narimatsu, H. Deletion polymorphism of SIGLEC14 and its functional implications. Glycobiology 2009, 19, 841–846. [Google Scholar] [CrossRef]
- Zou, F.; Gopalraj, R.K.; Lok, J.; Zhu, H.; Ling, I.F.; Simpson, J.F.; Tucker, H.M.; Kelly, J.F.; Younkin, S.G.; Dickson, D.W.; et al. Sex-dependent association of a common low-density lipoprotein receptor polymorphism with RNA splicing efficiency in the brain and Alzheimer’s disease. Hum. Mol. Genet. 2007, 17, 929–935. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, A.D.; Trowsdale, J. You say ITAM and I say ITIM, let’s call the whole thing off: The ambiguity of immunoreceptor signalling. Eur. J. Immunol. 2006, 36, 1646–1653. [Google Scholar] [CrossRef]
- Dushek, O.; Goyette, J.; van der Merwe, P.A. Non-catalytic tyrosine-phosphorylated receptors. Immunol. Rev. 2012, 250, 258–276. [Google Scholar] [CrossRef]
- Isakov, N. Immunoreceptor tyrosine-based activation motif (ITAM), a unique module linking antigen and Fc receptors to their signaling cascades. J. Leukoc. Biol. 1997, 61, 6–16. [Google Scholar] [CrossRef] [Green Version]
- Ravetch, J.V. Immune Inhibitory Receptors. Science 2000, 290, 84–89. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [CrossRef]
- Fishilevich, S.; Nudel, R.; Rappaport, N.; Hadar, R.; Plaschkes, I.; Iny Stein, T.; Rosen, N.; Kohn, A.; Twik, M.; Safran, M.; et al. GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, 2017. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, A.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef] [Green Version]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, J.A.; Woltjer, R.L.; Goodenbour, J.M.; Horvath, S.; Geschwind, D.H. Genes and pathways underlying regional and cell type changes in Alzheimer’s disease. Genome Med. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Melief, J.; Kooijman, L.; Huitinga, I.; Klooster, J.; Bossers, K.; Hol, E.M. Acute isolation and transcriptome characterization of cortical astrocytes and microglia from young and aged mice. Neurobiol. Aging 2014, 35, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wes, P.D.; Easton, A.; Corradi, J.; Barten, D.M.; Devidze, N.; Decarr, L.B.; Truong, A.; He, A.; Barrezueta, N.X.; Polson, C.; et al. Tau Overexpression Impacts a Neuroinflammation Gene Expression Network Perturbed in Alzheimer’s Disease. PLoS ONE 2014, 9, e106050. [Google Scholar] [CrossRef]
- Zhang, B.; Gaiteri, C.; Bodea, L.-G.; Wang, Z.; McElwee, J.; Podtelezhnikov, A.A.; Zhang, C.; Xie, T.; Tran, L.; Dobrin, R.; et al. Integrated Systems Approach Identifies Genetic Nodes and Networks in Late-Onset Alzheimer’s Disease. Cell 2013, 153, 707–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdollahi, M.R.; Huang, S.; Rodriguez, S.; Guthrie, P.A.I.; Smith, G.D.; Ebrahim, S.; Lawlor, D.A.; Day, I.N.M.; Gaunt, T.R. Homogeneous Assay of rs4343, anACEI/D Proxy, and an Analysis in the British Women’s Heart and Health Study (BWHHS). Dis. Markers 2008, 24, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Hinds, D.A.; Kloek, A.P.; Jen, M.; Chen, X.; Frazer, K.A. Common deletions and SNPs are in linkage disequilibrium in the human genome. Nat. Genet. 2006, 38, 82–85. [Google Scholar] [CrossRef]
- McCarroll, S.A.; Huett, A.; Kuballa, P.; Chilewski, S.D.; Landry, A.; Goyette, P.; Zody, M.C.; Hall, J.L.; Brant, S.R.; Cho, J.H.; et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat. Genet. 2008, 40, 1107–1112. [Google Scholar] [CrossRef] [PubMed]
- Machiela, M.J.; Chanock, S.J. LDlink: A web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Ariga, T.; McDonald, M.P.; Yu, R.K. Thematic Review Series: Sphingolipids. Role of ganglioside metabolism in the pathogenesis of Alzheimer’s disease—A review. J. Lipid Res. 2008, 49, 1157–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanagisawa, K. Role of gangliosides in Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Biomembr. 2007, 1768, 1943–1951. [Google Scholar] [CrossRef] [Green Version]
- Salminen, A.; Kaarniranta, K. Siglec receptors and hiding plaques in Alzheimer’s disease. J. Mol. Med. 2009, 87, 697–701. [Google Scholar] [CrossRef]
- Griciuc, A.; Patel, S.; Federico, A.N.; Choi, S.H.; Innes, B.J.; Oram, M.K.; Cereghetti, G.; McGinty, D.; Anselmo, A.; Sadreyev, R.I.; et al. TREM2 Acts Downstream of CD33 in Modulating Microglial Pathology in Alzheimer’s Disease. Neuron 2019, 103, 820–835.e827. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e1217. [Google Scholar] [CrossRef]
- Ebbert, M.T.W.; Jensen, T.D.; Jansen-West, K.; Sens, J.P.; Reddy, J.S.; Ridge, P.G.; Kauwe, J.S.K.; Belzil, V.; Pregent, L.; Carrasquillo, M.M.; et al. Systematic analysis of dark and camouflaged genes reveals disease-relevant genes hiding in plain sight. Genome. Biol. 2019, 20, 97. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | SNP | P (pQTL) | β (pQTL) | P (AD) | β (AD) | N (AD) | ITIM/ ITAM |
|---|---|---|---|---|---|---|---|
| CD33 | rs12459419 | 0 † | −0.94 | 7.13 × 10−9 | −0.01330 | 458,744 | ITIM |
| FCGR3B | rs10919543 | 3.20 × 10−67 | 0.44 | 0.000317 | 0.00806 | 445,293 | ITAM |
| LILRA5 | rs759819 | 2.50 × 10−111 | −0.54 | 0.00186 | 0.00717 | 454,216 | ITAM |
| LILRB2 | rs373032 | 7.60 × 10−146 | −0.72 | 0.00227 | 0.00763 | 463,880 | ITIM |
| SIGLEC9 | rs2075803 | 0 † | −1.23 | 0.00703 | 0.00576 | 466,252 | ITIM |
| SIRPB1 | rs3848788 | 1.20 × 10−213 | 0.75 | 0.00942 | 0.00582 | 458,092 | ITAM |
| COLEC12 | rs2846667 | 9.30 × 10−12 | 0.20 | 0.0177 | 0.00586 | 449,987 | ITAM |
| FCRL1 | rs4971155 | 6.30 × 10−26 | −0.26 | 0.0197 | −0.00520 | 403,829 | ITAM |
| NCR1 | rs2278428 | 1.10 × 10−15 | −0.36 | 0.0249 | 0.00815 | 466,252 | ITAM |
| SIGLEC14 | rs1106476 | 0 † | −1.19 | 0.0284 | 0.00736 | 458,063 | ITAM |
| FCRL3 | rs7528684 | 1.40 × 10−112 | 0.53 | 0.04 | −0.00434 | 458,744 | Both |
| MRC2 | rs146385050 | 1.30 × 10−11 | −0.22 | 0.041 | −0.00612 | 396,686 | ITAM |
| SLAMF6 | rs11291564 | 2.60 × 10−12 | 0.20 | 0.042 | −0.02450 | 17,477 | ITAM |
| pQTLs | ITIM/ITAM (%) | Not ITIM/ITAM (%) | Total |
|---|---|---|---|
| AD p < 0.05 | 13 (28) | 54 (10) | 67 |
| AD p > 0.05 | 34 (72) | 488 (90) | 522 |
| Total | 47 (100) | 542 (100) | 589 |
| SIGLEC14 Copies | rs1106476 T/T | rs1106476 A/T | rs1106476 A/A | Total |
|---|---|---|---|---|
| 0 | 0 | 1 | 1 | 2 |
| 1 | 6 | 13 | 0 | 19 |
| 2 | 39 | 0 | 0 | 39 |
| 3 | 2 | 2 | 0 | 4 |
| Total | 47 | 16 | 1 | 64 |
| SIGLEC14 Copy Number | Caucasian | African American | Other | Total |
|---|---|---|---|---|
| 0 | 24 | 74 | 44 | 142 |
| 1 | 304 | 348 | 316 | 968 |
| 2 | 692 | 522 | 652 | 1866 |
| 3 | 21 | 53 | 43 | 117 |
| 4 | 0 | 1 | 1 | 2 |
| Total | 1041 | 998 | 1056 | 3095 |
| Deletion MAF | 0.1691 | 0.2485 | 0.1913 | 0.2023 |
| Addition MAF | 0.0101 | 0.0276 | 0.0213 | 0.0195 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shaw, B.C.; Katsumata, Y.; Simpson, J.F.; Fardo, D.W.; Estus, S. Analysis of Genetic Variants Associated with Levels of Immune Modulating Proteins for Impact on Alzheimer’s Disease Risk Reveal a Potential Role for SIGLEC14. Genes 2021, 12, 1008. https://doi.org/10.3390/genes12071008
Shaw BC, Katsumata Y, Simpson JF, Fardo DW, Estus S. Analysis of Genetic Variants Associated with Levels of Immune Modulating Proteins for Impact on Alzheimer’s Disease Risk Reveal a Potential Role for SIGLEC14. Genes. 2021; 12(7):1008. https://doi.org/10.3390/genes12071008
Chicago/Turabian StyleShaw, Benjamin C., Yuriko Katsumata, James F. Simpson, David W. Fardo, and Steven Estus. 2021. "Analysis of Genetic Variants Associated with Levels of Immune Modulating Proteins for Impact on Alzheimer’s Disease Risk Reveal a Potential Role for SIGLEC14" Genes 12, no. 7: 1008. https://doi.org/10.3390/genes12071008
APA StyleShaw, B. C., Katsumata, Y., Simpson, J. F., Fardo, D. W., & Estus, S. (2021). Analysis of Genetic Variants Associated with Levels of Immune Modulating Proteins for Impact on Alzheimer’s Disease Risk Reveal a Potential Role for SIGLEC14. Genes, 12(7), 1008. https://doi.org/10.3390/genes12071008