
Retinoic Acid-Induced Gene G(RIG-G) as a Novel Monitoring Biomarker in Leukemia and Its Clinical Applications


 and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Specimens
2.2. Total RNA Extraction, Quantification of Total RNA and cDNA Synthesis
2.3. Primer and TaqMan Probe Design
2.4. Standard Curve Determination
2.5. Real-time PCR for RIG-G Gene mRNA
2.6. Agarose Gel Electrophoresis
2.7. Morphological Staining of Bone Marrow Cells (Wright’s Staining)
2.8. PML-RARα Fusion Gene Detection
2.9. Statistical Analysis
3. Results
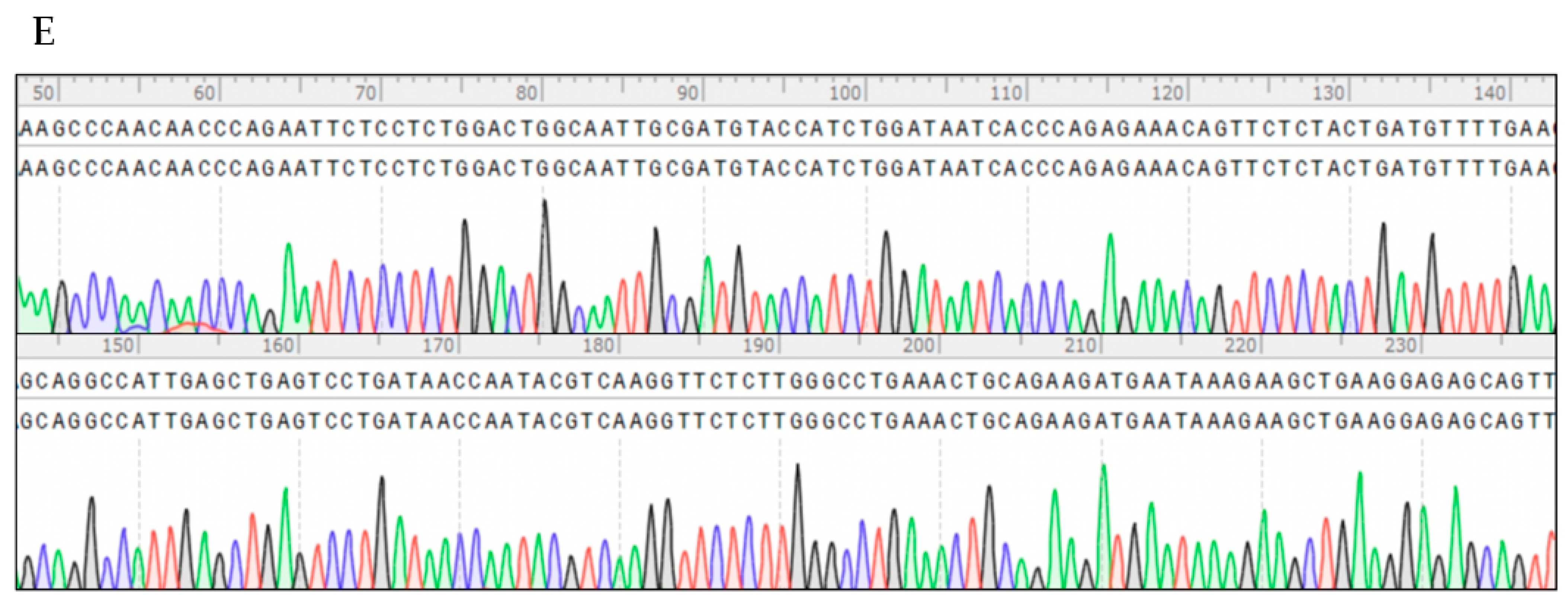
3.1. Development of TaqMan-MGB Real-Time Quantitative PCR Assay for RIG-G Gene
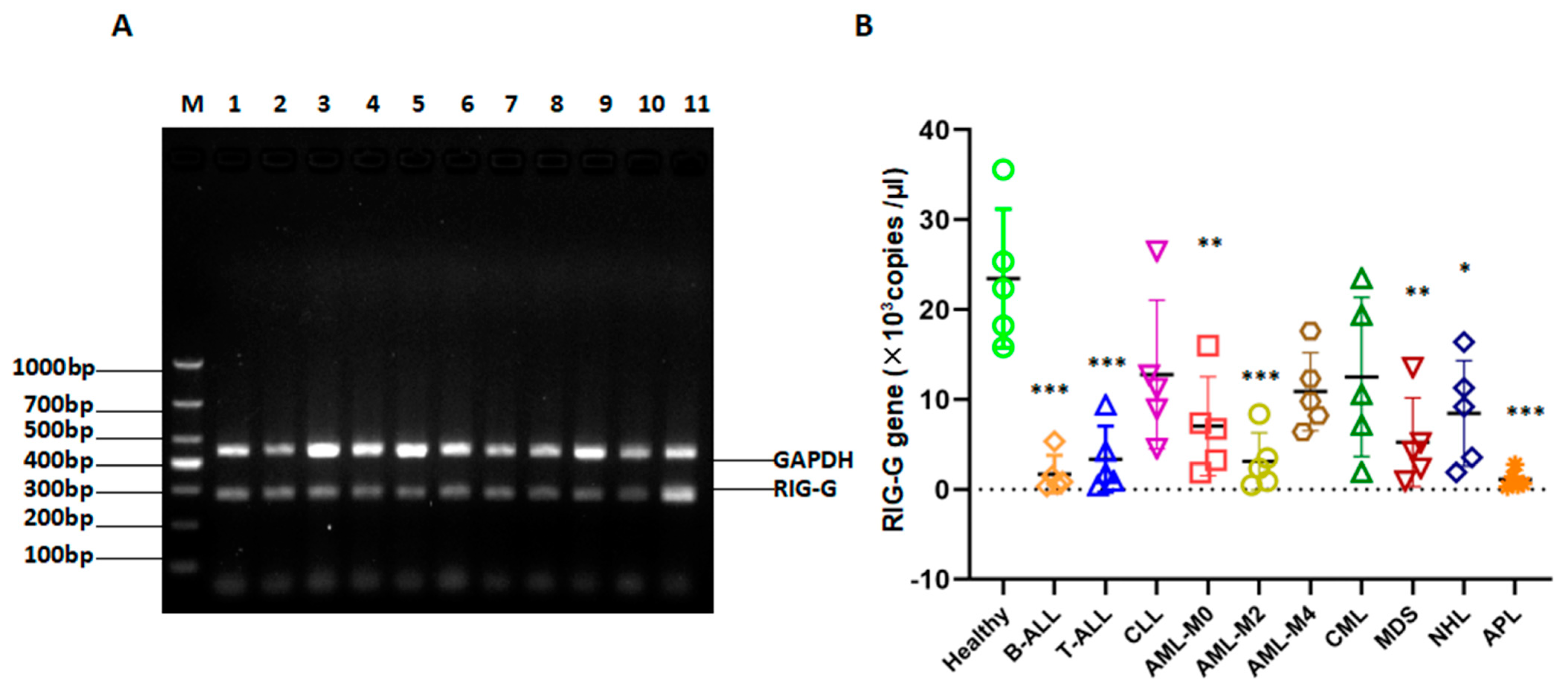
3.2. Methodological Performance Verification of the Established Method for RIG-G Gene Detection
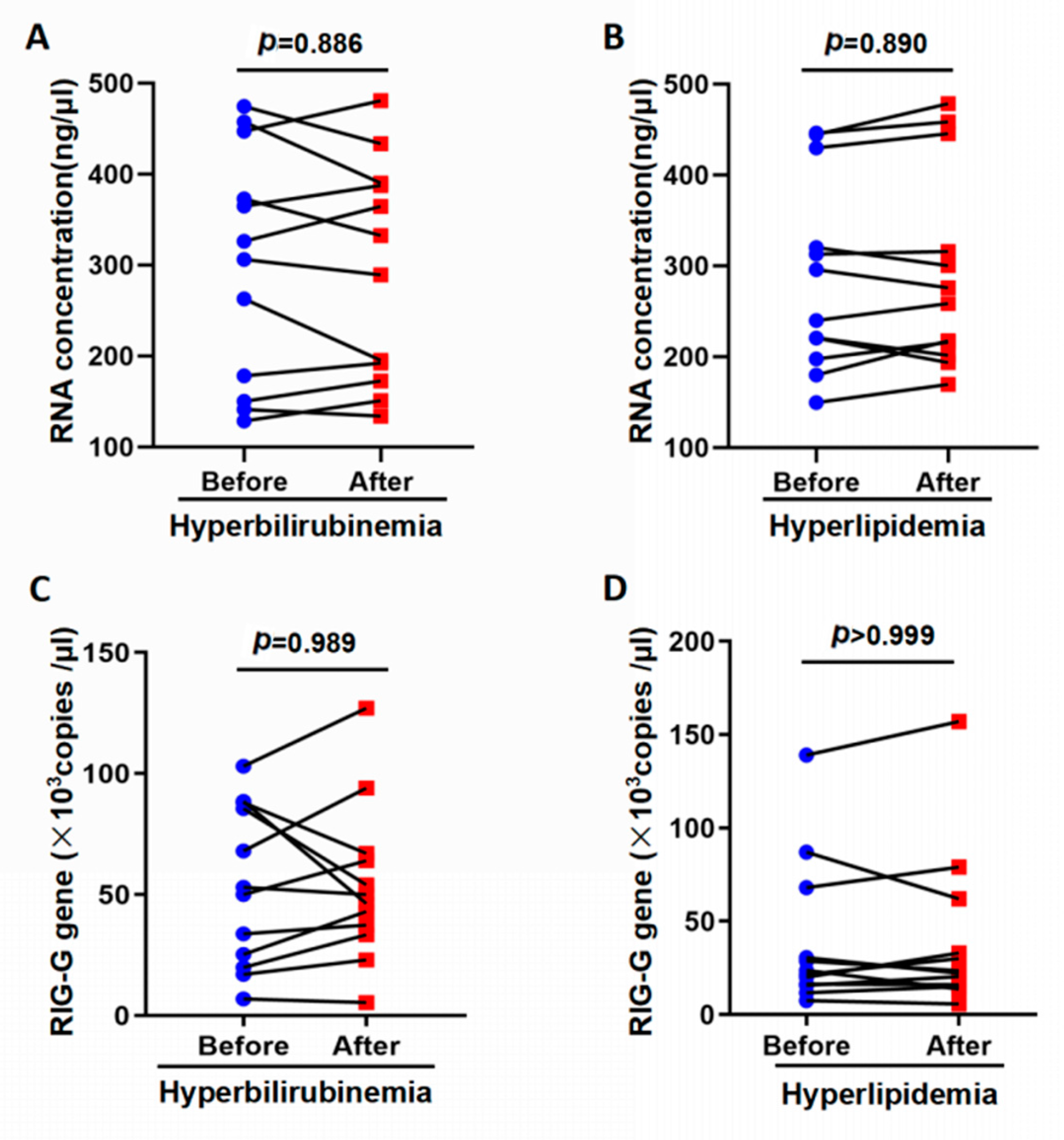
3.3. Influence of Interfering Substances on the Detection of RIG-G Gene
3.4. Evaluation of Established PCR Method for Detection of RIG-G mRNA in Peripheral Blood of APL Patients
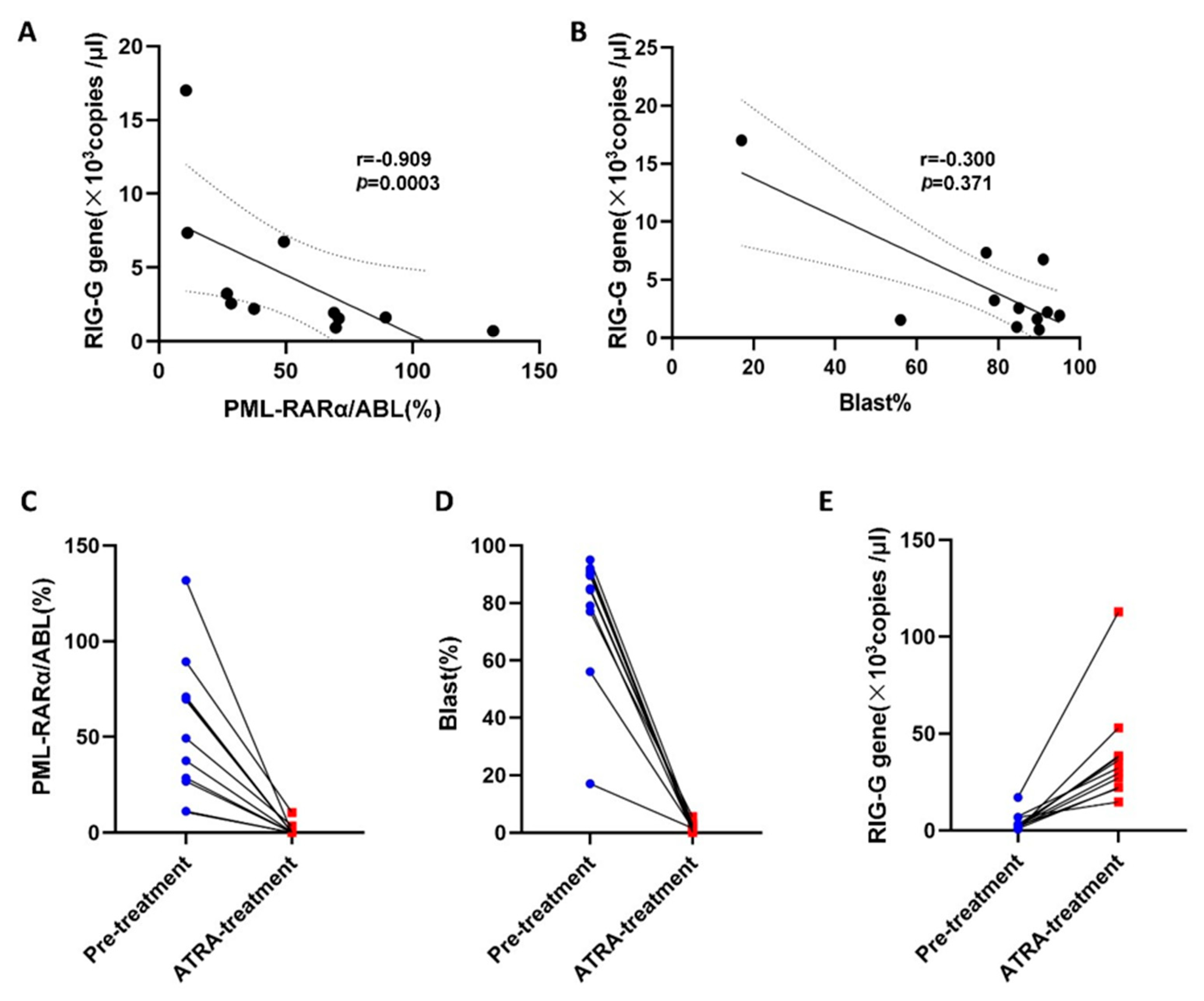
3.5. Relationship between RIG-G mRNA Expression and Prognosis in APL Patients
3.6. Correlation of RIG-G Gene Expression with PML-RARα Fusion Gene and Blast Cells in APL
3.7. RIG-G Gene Expression Is Reduced in Patients with Many Types of Leukemia
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Huang, M.E.; Ye, Y.C.; Chen, S.R.; Chai, J.-R.; Lu, J.-X.; Zhoa, L.; Gu, L.-J.; Wang, Z.-Y. Use of all-trans retinoic acid in the treatment of acute promyelocytic leukemia. Blood 1988, 72, 567–572. [Google Scholar] [CrossRef] [Green Version]
- Puccetti, E.; Ruthardt, M. Acute promyelocytic leukemia: PML/RARalpha and the leukemic stem cell. Leukemia 2004, 18, 1169–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tallmann, M.S. Curative therapeutic approaches to APL. Ann. Hematol. 2004, 83 (Suppl. S1), S81–S82. [Google Scholar] [PubMed]
- Chen, G.Q.; Shi, X.G.; Tang, W.; Xiong, S.M.; Zhu, J.; Cai, X.; Han, Z.G.; Ni, J.H.; Shi, G.Y.; Jia, P.M.; et al. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): I. As2O3 exerts dose-dependent dual effects on APL cells. Blood 1997, 89, 3345–3353. [Google Scholar] [PubMed]
- O’Donnell, M.R.; Tallman, M.S.; Abboud, C.N.; Altman, J.K.; Appelbaum, F.R.; Arber, D.A.; Attar, E.; Borate, U.; Coutre, S.E.; Damon, L.E.; et al. Acute Myeloid Leukemia, Version 2.2013. J. Natl. Compr. Cancer Netw. 2013, 11, 1047–1055. [Google Scholar] [CrossRef] [Green Version]
- Seftel, M.; Barnett, M.; Couban, S.; Leber, B.; Storring, J.; Assaily, W.; Fuerth, B.; Christofides, A.; Schuh, A. A Canadian Consensus on the Management of Newly Diagnosed and Relapsed Acute Promyelocytic Leukemia in Adults. Curr. Oncol. 2014, 21, 234–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Govind Babu, K.; Lokesh, K.N.; Suresh Babu, M.C.; Bhat, G.R. Acute coronary syndrome manifesting as an adverse effect of all-trans-retinoic acid in acute promyelocytic leukemia: A case report with review of the literature and a spotlight on man-agement. Case Rep. Oncol. Med. 2016, 2016, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yu, M.; Tong, J.H.; Mao, M.; Kan, L.-X.; Liu, M.-M.; Sun, Y.-W.; Fu, G.; Jing, Y.-K.; Yu, L.; Lepaslier, D.; et al. Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute pro-myelocytic leukemia cells andrepresenting a new member of a family of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 1997, 94, 7406–7411. [Google Scholar] [CrossRef] [Green Version]
- Xiao, S.; Li, D.; Zhu, H.-Q.; Song, M.-G.; Pan, X.-R.; Jia, P.-M.; Peng, L.-L.; Dou, A.-X.; Chen, G.-Q.; Chen, S.-J.; et al. RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc. Natl. Acad. Sci. USA 2006, 103, 16448–16453. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.P.; Zhang, Z.L.; Xiao, S.; Zhuang, L.-K.; Xia, D.; Zou, Q.-P.; Jia, P.-M.; Tong, J.-H. Rig-G negatively regulates SCF-E3 ligase activities by disrupting the assembly of COP9 signal-osome complex. Biochem. Biophys. Res. Commun. 2013, 432, 425–430. [Google Scholar] [CrossRef]
- Leeflang, M.M.; Allerberger, F. How to: Evaluate a diagnostic test. Clin. Microbiol. Infect. 2019, 25, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Eusebi, P. Diagnostic Accuracy Measures. Cerebrovasc. Dis. 2013, 36, 267–272. [Google Scholar] [CrossRef]
- Sun, J.; Wang, X.; Liu, W.; Ji, P.; Shang, A.; Wu, J.; Zhou, H.; Quan, W.; Yao, Y.; Yang, Y.; et al. Novel evidence for retinoic acid-induced G (Rig-G) as a tumor suppressor by activating p53 signaling pathway in lung cancer. FASEB J. 2020, 34, 11900–11912. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, J.; Korístek, Z.; Starý, J.; Zák, P.; Kozák, T.; Marková, J.; Michalová, K.; Dvoráková, D.; Mayer, J.; Cetkovský, P. Therapy of acute promyelocytic leukemia in Czechia: Results and analysis of prognostic factors. Vnitrni Lek. 2008, 54, 757–770. [Google Scholar]
- Lou, Y.J.; Pan, X.R.; Jia, P.M.; Li, D.; Xiao, S.; Zhang, Z.-L.; Chen, S.-J.; Chen, Z.; Tong, J.-H. IRF-9/STAT2 [corrected] functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res. 2009, 69, 3673–3680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lou, Y.-J.; Zhang, Z.-L.; Pan, X.-R.; Xu, G.-P.; Jia, P.-M.; Li, N.; Tong, J.-H. Intact JAK–STAT signaling pathway is a prerequisite for STAT1 to reinforce the expression of RIG-G gene. Exp. Cell Res. 2011, 317, 513–520. [Google Scholar] [CrossRef]
- Gu, Z.-M.; Liu, C.-X.; Wu, S.-F.; Zhao, M.; Xu, H.-Z.; Liu, W.; Zhou, H.-C.; Chen, G.-Q.; Wu, Y.-L. PU.1 directly regulates retinoic acid-induced expression of RIG-G in leukemia cells. FEBS Lett. 2010, 585, 375–380. [Google Scholar] [CrossRef] [Green Version]
- Golenberg, E.M.; Bickel, A.; Weihs, P. Effect of Highly Fragmented DNA on PCR. Nucleic Acids Res. 1996, 24, 5026–5033. [Google Scholar] [CrossRef] [Green Version]
- Köchl, S.; Niederstätter, H.; Parson, W. DNA Extraction and Quantitation of Forensic Samples Using the Phenol–Chloroform Method and Real-Time PCR. Forensic DNA Typing Protoc. 2004, 297, 13–30. [Google Scholar]
- Chen, S.C.; Halliday, C.L.; Meyer, W. A review of nucleic acid-based diagnostic tests for systemic mycoses with an emphasis on polymerase chain reaction-based assays. Med. Mycol. 2002, 40, 333–357. [Google Scholar] [CrossRef]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors - occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Fischer, S.; Keshavjee, S.; Liu, M. Heparin interference with reverse transcriptase polymerase chain reaction of RNA extracted from lungs after ischemia-reperfusion. Transpl. Int. 2000, 13, 146–150. [Google Scholar] [CrossRef]
- Zhang, Z.; Kermekchiev, M.B.; Barnes, W.M. Direct DNA amplification from crude clinical samples using a PCR enhancer cocktail and novelmutants of Taq. J. Mol. Diagn. 2010, 12, 1521–1561. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Liu, J.; Kuang, X.; Dai, M.; Cheng, G.; Wang, X.; Peng, D.; Huang, L.; Ahmad, I.; Ren, N.; et al. WITHDRAWN: Identification of Campylobacter jejuni and determination of point mutations associated with macrolide resistance using a multiplex TaqMan MGB real-time PCR. J. Glob. Antimicrob. Resist. 2014, 118, 1418–1425. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene/Probe | Primer Sequence |
|---|---|
| RIG-G | F: 5′-GAAGAAATGAAAGGGCGAAGG-3′ |
| R: 5′-AGGACATCTGTTTGGCAAGGAG-3′ | |
| GAPDH | F: 5′- AGGTCGGAGTCAACGGATTTGGT-3′ |
| R: 5′- GTGCAGGAGGCATTGCTGATGAT-3′ | |
| Taqman RIG-G-probe | 5′-FAM-AGGACTCAGCTCAATGG-MGB-3′ |
| Assay | Gold Standard | Sum Total | ||
|---|---|---|---|---|
| APL(+) | APL(-) | |||
| RIG-G gene | High | 17 | 0 | 17 |
| Low | 3 | 40 | 43 | |
| Sum total | 20 | 40 | 60 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, F.; Tian, J.; Pang, L.; Wu, J.; Shang, A.; Sun, Z.; Li, D.; Yan, J.; Quan, W. Retinoic Acid-Induced Gene G(RIG-G) as a Novel Monitoring Biomarker in Leukemia and Its Clinical Applications. Genes 2021, 12, 1035. https://doi.org/10.3390/genes12071035
Wang F, Tian J, Pang L, Wu J, Shang A, Sun Z, Li D, Yan J, Quan W. Retinoic Acid-Induced Gene G(RIG-G) as a Novel Monitoring Biomarker in Leukemia and Its Clinical Applications. Genes. 2021; 12(7):1035. https://doi.org/10.3390/genes12071035
Chicago/Turabian StyleWang, Fei, Jiale Tian, Li Pang, Junlu Wu, Anquan Shang, Zujun Sun, Dong Li, Jinsong Yan, and Wenqiang Quan. 2021. "Retinoic Acid-Induced Gene G(RIG-G) as a Novel Monitoring Biomarker in Leukemia and Its Clinical Applications" Genes 12, no. 7: 1035. https://doi.org/10.3390/genes12071035