
A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Identification of Genes Associated with AMC
2.2. Identification of Genes Associated with Scoliosis
2.3. Identification of Genes Associated with Both AMC and Scoliosis and Gene Interaction Pathway Analysis
2.4. Identification of Copy Number Variants (CNV) Associated with AMC and Scoliosis
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hall, J.G. Arthrogryposis (multiple congenital contractures): Diagnostic approach to etiology, classification, genetics, and general principles. Eur. J. Med. Genet. 2014, 57, 464–472. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G.; Kiefer, J. Arthrogryposis as a Syndrome: Gene Ontology Analysis. Mol. Syndr. 2016, 7, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, J.; Hall, J.G. Gene ontology analysis of arthrogryposis (multiple congenital contractures). Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 310–326. [Google Scholar] [CrossRef]
- Bamshad, M.; Van Heest, A.; Pleasure, D. Arthrogryposis: A Review and Update. J. Bone Jt. Surg. Am. 2009, 91, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.G.; Kimber, E.; Dieterich, K. Classification of arthrogryposis. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 300–303. [Google Scholar] [CrossRef]
- Hall, J.G.; Aldinger, K.A.; Tanaka, K.I. Amyoplasia revisited. Am. J. Med. Genet. A 2014, 164, 700–730. [Google Scholar] [CrossRef]
- Drummond, D.S.; A Mackenzie, D. Scoliosis in Arthrogryposis Multiplex Congenita. Spine 1978, 3, 146–151. [Google Scholar] [CrossRef]
- Herron, L.D.; Westin, G.W.; Dawson, E.G. Scoliosis in arthrogryposis multiplex congenita. J. Bone Jt. Surg. Am. 1978, 60, 293–299. [Google Scholar] [CrossRef]
- Komolkin, I.; Ulrich, E.V.; Agranovich, O.E.; van Bosse, H.J. Treatment of Scoliosis Associated with Arthrogryposis Multiplex Congenita. J. Pediatr. Orthop. 2017, 37, S24–S26. [Google Scholar] [CrossRef]
- Yingsakmongkol, W.; Kumar, S.J. Scoliosis in arthrogryposis multiplex congenita: Results after nonsurgical and surgical treatment. J. Pediatr. Orthop. 2000, 20, 656–661. [Google Scholar] [CrossRef]
- Cheng, J.C.; Castelein, R.M.; Chu, W.C.; Danielsson, A.J.; Dobbs, M.B.; Grivas, T.B.; Gurnett, C.A.; Luk, K.D.; Moreau, A.; Newton, P.O.; et al. Adolescent idiopathic scoliosis. Nat. Rev. Dis. Primers 2015, 1, 15030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Machado, G.; Berenguer-Pascual, E.; Bovea-Marco, M.; Rubio-Belmar, P.A.; García-López, E.; Garzón, M.J.G.; Mena-Mollá, S.; Pallardó, F.V.; Bas, T.; Viña, J.R.; et al. From genetics to epigenetics to unravel the etiology of adolescent idiopathic scoliosis. Bone 2020, 140, 115563. [Google Scholar] [CrossRef] [PubMed]
- Baer, S.; Obringer, C.; Julia, S.; Chelly, J.; Capri, Y.; Gras, D.; Baujat, G.; Felix, T.M.; Doray, B.; Del Pozo, J.S.; et al. Early-onset nucleotide excision repair disorders with neurological impairment: Clues for early diagnosis and prognostic counseling. Clin. Genet. 2020, 98, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pearce, M.P.; Sliter, D.A.; Olzmann, J.A.; Christianson, J.C.; Kopito, R.R.; Boeckmann, S.; Gagen, C.; Leichner, G.S.; Roitelman, J.; et al. SPFH1 and SPFH2 mediate the ubiquitination and degradation of inositol 1,4,5-trisphosphate receptors in muscarinic receptor-expressing HeLa cells. Biochim. Biophys. Acta BBA Bioenerg. 2009, 1793, 1710–1718. [Google Scholar] [CrossRef] [Green Version]
- Pelin, K.; Hilpelä, P.; Donner, K.; Sewry, C.; Akkari, P.A.; Wilton, S.D.; Wattanasirichaigoon, D.; Bang, M.-L.; Centner, T.; Hanefeld, F.; et al. Mutations in the nebulin gene associated with autosomal recessive nemaline myopathy. Proc. Natl. Acad. Sci. USA 1999, 96, 2305–2310. [Google Scholar] [CrossRef] [Green Version]
- Laing, N.G.; Dye, D.E.; Wallgren-Pettersson, C.; Richard, G.; Monnier, N.; Lillis, S.; Winder, T.L.; Lochmuller, H.; Graziano, C.; Mitrani-Rosenbaum, S.; et al. Mutations and polymorphisms of the skeletal muscle alpha-actin gene (ACTA1). Hum. Mutat. 2009, 30, 1267–1277. [Google Scholar] [CrossRef] [Green Version]
- Somerville, R.P.; Jungers, K.A.; Apte, S.S. Discovery and characterization of a novel, widely expressed metalloprotease, ADAMTS10, and its proteolytic activation. J. Biol. Chem. 2004, 279, 51208–51217. [Google Scholar] [CrossRef] [Green Version]
- Cappello, S.; Gray, M.J.; Badouel, C.; Lange, S.; Einsiedler, M.; Srour, M.; Chitayat, D.; Hamdan, F.F.; Jenkins, Z.A.; Morgan, T.R.; et al. Mutations in genes encoding the cadherin receptor-ligand pair DCHS1 and FAT4 disrupt cerebral cortical development. Nat. Genet. 2013, 45, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Foldynova-Trantirkova, S.; Wilcox, W.R.; Krejci, P. Sixteen years and counting: The current understanding of fibroblast growth factor receptor 3 (FGFR3) signaling in skeletal dysplasias. Hum. Mutat. 2012, 33, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Hellmann, T.V.; Nickel, J.; Mueller, T.D. Missense Mutations in GDF-5 Signaling: Molecular Mechanisms behind Skeletal Malformation. In Mutations in Human Genetic Disease; David, D., Chen, J.-M., Eds.; IntechOpen: Wuerzburg, Germany, 2012; Volume 1, pp. 1–45. [Google Scholar]
- Bai, X.; Xiao, Z.; Pan, Y.; Hu, J.; Pohl, J.; Wen, J.; Li, L. Cartilage-derived morphogenetic protein-1 promotes the differentiation of mesenchymal stem cells into chondrocytes. Biochem. Biophys. Res. Commun. 2004, 325, 453–460. [Google Scholar] [CrossRef]
- Quélin, C.; Loget, P.; Rozel, C.; D’Hervé, D.; Fradin, M.; Demurger, F.; Odent, S.; Pasquier, L.; Cavé, H.; Marcorelles, P. Fetal costello syndrome with neuromuscular spindles excess and p.Gly12Val HRAS mutation. Eur. J. Med. Genet. 2017, 60, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Wassif, C.A.; Maslen, C.; Kachilele-Linjewile, S.; Lin, D.; Linck, L.M.; Connor, W.E.; Steiner, R.D.; Porter, F.D. Mutations in the human sterol delta7-reductase gene at 11q12-13 cause Smith-Lemli-Opitz syndrome. Am. J. Hum. Genet. 1998, 63, 55–62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kishino, T.; Lalande, M.; Wagstaff, J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat. Genet. 1997, 15, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Basson, C.T.; Huang, T.; Lin, R.C.; Bachinsky, D.R.; Weremowicz, S.; Vaglio, A.; Bruzzone, R.; Quadrelli, R.; Lerone, M.; Romeo, G.; et al. Different TBX5 interactions in heart and limb defined by Holt-Oram syndrome mutations. Proc. Natl. Acad. Sci. USA 1999, 96, 2919–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasson, P.; DeLaurier, A.; Bennett, M.; Grigorieva, E.; Naiche, L.; Papaioannou, V.; Mohun, T.J.; Logan, M.P. Tbx4 and Tbx5 Acting in Connective Tissue Are Required for Limb Muscle and Tendon Patterning. Dev. Cell 2010, 18, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Davignon, L.; Chauveau, C.; Julien, C.; Dill, C.; Duband-Goulet, I.; Cabet, E.; Buendia, B.; Lilienbaum, A.; Rendu, J.; Minot, M.C.; et al. The transcription coactivator ASC-1 is a regulator of skeletal myogenesis, and its deficiency causes a novel form of congenital muscle disease. Hum. Mol. Genet. 2016, 25, 1559–1573. [Google Scholar] [CrossRef] [Green Version]
- Birouk, N.; Azzedine, H.; Dubourg, O.; Muriel, M.-P.; Benomar, A.; Hamadouche, T.; Maisonobe, T.; Ouazzani, R.; Brice, A.; Yahyaoui, M.; et al. Phenotypical Features of a Moroccan Family With Autosomal Recessive Charcot-Marie-Tooth Disease Associated With the S194X Mutation in the GDAP1 Gene. Arch. Neurol. 2003, 60, 598–604. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.W.; Kim, S.M.; Sunwoo, I.N.; Cho, S.Y.; Hwang, S.J.; Kim, J.; Kang, S.H.; Park, K.-D.; Choi, K.-G.; Choi, I.S.; et al. A novel GDAP1 Q218E mutation in autosomal dominant Charcot-Marie-Tooth disease. J. Hum. Genet. 2008, 53, 360–364. [Google Scholar] [CrossRef] [Green Version]
- Nelis, E.; Erdem, S.; Bergh, P.Y.V.D.; Belpaire-Dethiou, M.-C.; Ceuterick, C.; Van Gerwen, V.; Cuesta, A.; Pedrola, L.; Palau, F.; Gabreels-Festen, A.A.; et al. Mutations in GDAP1: Autosomal recessive CMT with demyelination and axonopathy. Neurology 2002, 59, 1865–1872. [Google Scholar] [CrossRef]
- Baxter, R.V.; Ben Othmane, K.; Rochelle, J.M.; Stajich, J.; Hulette, C.; Dew-Knight, S.; Hentati, F.; Ben Hamida, M.; Bel, S.; Stenger, J.E.; et al. Ganglioside-induced differentiation-associated protein-1 is mutant in Charcot-Marie-Tooth disease type 4A/8q21. Nat. Genet. 2001, 30, 21–22. [Google Scholar] [CrossRef]
- Cuesta, A.; Pedrola, L.; Sevilla, T.; García-Planells, J.; Chumillas, M.J.; Mayordomo, F.; LeGuern, E.; Marín, I.; Vílchez, J.J.; Palau, F. The gene encoding ganglioside-induced differentiation-associated protein 1 is mutated in axonal Charcot-Marie-Tooth type 4A disease. Nat. Genet. 2001, 30, 22–25. [Google Scholar] [CrossRef]
- Ridanpää, M.; van Eenennaam, H.; Pelin, K.; Chadwick, R.; Johnson, C.; Yuan, B.; Vanvenrooij, W.; Pruijn, G.; Salmela, R.; Rockas, S.; et al. Mutations in the RNA Component of RNase MRP Cause a Pleiotropic Human Disease, Cartilage-Hair Hypoplasia. Cell 2001, 104, 195–203. [Google Scholar] [CrossRef] [Green Version]
- Rosenbluh, J.; Nijhawan, D.; Chen, Z.; Wong, K.-K.; Masutomi, K.; Hahn, W.C. RMRP Is a Non-Coding RNA Essential for Early Murine Development. PLoS ONE 2011, 6, e26270. [Google Scholar] [CrossRef] [Green Version]
- Morgan, N.; Brueton, L.A.; Cox, P.; Greally, M.T.; Tolmie, J.; Pasha, S.; Aligianis, I.A.; van Bokhoven, J.; Marton, T.; Al-Gazali, L.; et al. Mutations in the Embryonal Subunit of the Acetylcholine Receptor (CHRNG) Cause Lethal and Escobar Variants of Multiple Pterygium Syndrome. Am. J. Hum. Genet. 2006, 79, 390–395. [Google Scholar] [CrossRef] [Green Version]
- McMillin, M.J.; Beck, A.E.; Chong, J.X.; Shively, K.M.; Buckingham, K.J.; Gildersleeve, H.I.; Aracena, M.I.; Aylsworth, A.S.; Bitoun, P.; Carey, J.C.; et al. Mutations in PIEZO2 cause Gordon syndrome, Marden-Walker syndrome, and distal arthrogryposis type 5. Am. J. Hum. Genet. 2014, 94, 734–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cortez, D.; Guntuku, S.; Qin, J.; Elledge, S.J. ATR and ATRIP: Partners in Checkpoint Signaling. Science 2001, 294, 1713–1716. [Google Scholar] [CrossRef]
- Mokrani-Benhelli, H.; Gaillard, L.; Biasutto, P.; Le Guen, T.; Touzot, F.; Vasquez, N.; Komatsu, J.; Conseiller, E.; Picard, C.; Gluckman, E.; et al. Primary microcephaly, impaired DNA replication, and genomic instability caused by compound heterozygous ATR mutations. Hum. Mutat. 2013, 34, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Bajayo, A.; Goshen, I.; Feldman, S.; Csernus, V.; Iverfeldt, K.; Shohami, E.; Yirmiya, R.; Bab, I. Central IL-1 receptor signaling regulates bone growth and mass. Proc. Natl. Acad. Sci. USA 2005, 102, 12956–12961. [Google Scholar] [CrossRef] [Green Version]
- Wade, E.M.; Daniel, P.B.; Jenkins, Z.A.; McInerney-Leo, A.; Leo, P.; Morgan, T.; Addor, M.C.; Adès, L.C.; Bertola, D.; Bohring, A.; et al. Mutations in MAP3K7 that Alter the Activity of the TAK1 Signaling Complex Cause Frontometaphyseal Dysplasia. Am. J. Hum. Genet. 2016, 99, 392–406. [Google Scholar] [CrossRef] [Green Version]
- Matanis, T.; Akhmanova, A.; Wulf, P.S.; Del Nery, E.; Weide, T.; Stepanova, T.; Galjart, N.; Grosveld, F.; Goud, B.; De Zeeuw, C.I.; et al. Bicaudal-D regulates COPI-independent Golgi–ER transport by recruiting the dynein–dynactin motor complex. Nat. Cell Biol. 2002, 4, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Oates, E.C.; Rossor, A.M.; Hafezparast, M.; Gonzalez, M.; Speziani, F.; MacArthur, D.G.; Lek, M.; Cottenie, E.; Scoto, M.; Foley, A.R.; et al. Mutations in BICD2 Cause Dominant Congenital Spinal Muscular Atrophy and Hereditary Spastic Paraplegia. Am. J. Hum. Genet. 2013, 92, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nousiainen, H.; Kestilä, M.; Pakkasjärvi, N.; Honkala, H.; Kuure, S.; Tallila, J.; Vuopala, K.; Ignatius, J.; Herva, R.; Peltonen, L. Mutations in mRNA export mediator GLE1 result in a fetal motoneuron disease. Nat. Genet. 2008, 40, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Gruber, A.; Cardoso, M.; Taipa, R.; Fineza, I.; Gonçalves, A.; Laner, A.; Winder, T.L.; Schroeder, J.; Rath, J.; et al. LAMA2gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum. Mutat. 2018, 39, 1314–1337. [Google Scholar] [CrossRef] [PubMed]
- Toydemir, R.M.; Bamshad, M.J. Sheldon-Hall syndrome. Orphanet J. Rare Dis. 2009, 4, 11–15. [Google Scholar] [CrossRef] [Green Version]
- Ansari, M.; Rainger, J.K.; Murray, J.E.; Hanson, I.; Firth, H.V.; Mehendale, F.; Amiel, J.; Gordon, C.; Percesepe, A.; Mazzanti, L.; et al. A syndromic form of Pierre Robin sequence is caused by 5q23 deletions encompassing FBN2 and PHAX. Eur. J. Med. Genet. 2014, 57, 587–595. [Google Scholar] [CrossRef] [Green Version]
- Huang, N.; Lee, I.; Marcotte, E.M.; Hurles, M.E. Characterising and Predicting Haploinsufficiency in the Human Genome. PLoS Genet. 2010, 6, e1001154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Callewaert, B.; Loeys, B.; Ficcadenti, A.; Vermeer, S.; Landgren, M.; Kroes, H.Y.; Yaron, Y.; Pope, M.; Foulds, N.; Boute, O.; et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: Report of 14 novel mutations and review of the literature. Hum. Mutat. 2009, 30, 334–341. [Google Scholar] [CrossRef]
- Kragesteen, B.K.; Brancati, F.; Digilio, M.C.; Mundlos, S.; Spielmann, M. H2AFY promoter deletion causes PITX1 endoactivation and Liebenberg syndrome. J. Med. Genet. 2019, 56, 246–251. [Google Scholar] [CrossRef]
- Frints, S.G.; Hennig, F.; Colombo, R.; Jacquemont, S.; Terhal, P.; Zimmerman, H.H.; Hunt, D.; Mendelsohn, B.A.; Kordaß, U.; Webster, R.; et al. Deleterious de novo variants of X-linked ZC4H2 in females cause a variable phenotype with neurogenic arthrogryposis multiplex congenita. Hum. Mutat. 2019, 40, 2270–2285. [Google Scholar] [CrossRef] [Green Version]
- Rao, D.; Kronert, W.A.; Guo, Y.; Hsu, K.H.; Sarsoza, F.; Bernstein, S.I. Reductions in ATPase activity, actin sliding velocity, and myofibril stability yield muscle dysfunction in Drosophila models of myosin-based Freeman–Sheldon syndrome. Mol. Biol. Cell 2019, 30, 30–41. [Google Scholar] [CrossRef]
- Watson, C.M.; A Crinnion, L.; Murphy, H.; Newbould, M.; Harrison, S.M.; Lascelles, C.; Antanaviciute, A.; Carr, I.M.; Sheridan, E.; Bonthron, D.; et al. Deficiency of the myogenic factor MyoD causes a perinatally lethal fetal akinesia. J. Med. Genet. 2016, 53, 264–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, M.; Rigby, P.W. Gene Regulatory Networks and Transcriptional Mechanisms that Control Myogenesis. Dev. Cell 2014, 28, 225–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, C.; Li, Z.; Ramanujan, K.; Clay, I.; Zhang, Y.; Lemire-Brachat, S.; Glass, D.J. A Long Non-coding RNA, LncMyoD, Regulates Skeletal Muscle Differentiation by Blocking IMP2-Mediated mRNA Translation. Dev. Cell 2015, 34, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gicquel, C.; Rossignol, S.; Cabrol, S.; Houang, M.; Steunou, V.; Barbu, V.; Danton, F.; Thibaud, N.; Le Merrer, M.; Burglen, L.; et al. Epimutation of the telomeric imprinting center region on chromosome 11p15 in Silver-Russell syndrome. Nat. Genet. 2005, 37, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Bruce, S.; Hannula-Jouppi, K.; Peltonen, J.; Kere, J.; Lipsanen-Nyman, M. Clinically Distinct Epigenetic Subgroups in Silver-Russell Syndrome: The Degree ofH19Hypomethylation Associates with Phenotype Severity and Genital and Skeletal Anomalies. J. Clin. Endocrinol. Metab. 2009, 94, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Saal, H.M.; Harbison, M.D.; Netchine, I. Silver-Russell Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Meerschaut, I.; De Coninck, S.; Steyaert, W.; Barnicoat, A.; Bayat, A.; Benedicenti, F.; Berland, S.; Blair, E.M.; Breckpot, J.; De Burca, A.; et al. A clinical scoring system for congenital contractural arachnodactyly. Genet. Med. 2020, 22, 124–131. [Google Scholar] [CrossRef]
- Alvarado, D.M.; McCall, K.; Aferol, H.; Silva, M.J.; Garbow, J.R.; Spees, W.M.; Patel, T.; Siegel, M.; Dobbs, M.B.; Gurnett, C.A. Pitx1 haploinsufficiency causes clubfoot in humans and a clubfoot-like phenotype in mice. Hum. Mol. Genet. 2011, 20, 3943–3952. [Google Scholar] [CrossRef] [Green Version]
- Hirata, H.; Nanda, I.; van Riesen, A.; McMichael, G.; Hu, H.; Hambrock, M.; Papon, M.-A.; Fischer, U.; Marouillat, S.; Ding, C.; et al. ZC4H2 Mutations Are Associated with Arthrogryposis Multiplex Congenita and Intellectual Disability through Impairment of Central and Peripheral Synaptic Plasticity. Am. J. Hum. Genet. 2013, 92, 681–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- May, M.; Hwang, K.-S.; Miles, J.; Williams, C.; Niranjan, T.; Kahler, S.G.; Chiurazzi, P.; Steindl, K.; Van Der Spek, P.J.; Swagemakers, S.; et al. ZC4H2, an XLID gene, is required for the generation of a specific subset of CNS interneurons. Hum. Mol. Genet. 2015, 24, 4848–4861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanzottera, C.; Milani, D.; Alfei, E.; Rizzo, A.; D’Arrigo, S.; Esposito, S.; Pantaleoni, C. ZC4H2 deletions can cause severe phenotype in female carriers. Am. J. Med. Genet. Part A 2017, 173, 1358–1363. [Google Scholar] [CrossRef]
- Vogt, J.; Morgan, N.V.; Rehal, P.; Faivre, L.; Brueton, L.A.; Becker, K.; Fryns, J.P.; Holder, S.; Islam, L.; Kivuva, E.; et al. CHRNG genotype-phenotype correlations in the multiple pterygium syndromes. J. Med. Genet. 2012, 49, 21–26. [Google Scholar] [CrossRef]
- Gurnett, C.A.; Desruisseau, D.M.; McCall, K.; Choi, R.; Meyer, Z.I.; Talerico, M.; Miller, S.E.; Ju, J.-S.; Pestronk, A.; Connolly, A.M.; et al. Myosin binding protein C1: A novel gene for autosomal dominant distal arthrogryposis type 1. Hum. Mol. Genet. 2010, 19, 1165–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, K.; Buchan, J.G.; Alvarado, D.M.; McCall, K.; Vydyanath, A.; Luther, P.; Goldsmith, M.I.; Dobbs, M.B.; Gurnett, C.A. MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis. Hum. Mol. Genet. 2013, 22, 4967–4977. [Google Scholar] [CrossRef] [Green Version]
- Cameron-Christie, S.R.; Wells, C.F.; Simon, M.; Wessels, M.; Tang, C.Z.; Wei, W.; Takei, R.; Aarts-Tesselaar, C.; Sandaradura, S.; Sillence, D.O.; et al. Recessive Spondylocarpotarsal Synostosis Syndrome Due to Compound Heterozygosity for Variants in MYH3. Am. J. Hum. Genet. 2018, 102, 1115–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chong, J.X.; Burrage, L.C.; Beck, A.E.; Marvin, C.T.; McMillin, M.J.; Shively, K.M.; Harrell, T.M.; Buckingham, K.J.; Bacino, C.A.; Jain, M.; et al. Autosomal-Dominant Multiple Pterygium Syndrome Is Caused by Mutations in MYH3. Am. J. Hum. Genet. 2015, 96, 841–849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittle, J.; Antunes, L.; Harris, M.; Upshaw, Z.; Sepich, D.S.; Johnson, A.N.; Mokalled, M.; Solnica-Krezel, L.; Dobbs, M.B.; A Gurnett, C. MYH 3-associated distal arthrogryposis zebrafish model is normalized with para-aminoblebbistatin. EMBO Mol. Med. 2020, 12, e12356. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, F.; Zhao, Y.; Yang, P.; Chen, J.; Sun, H.; Liu, L.; Li, W.; Pan, L.; Guo, Y.; et al. A Gain-of-Function Mutation in Tnni2 Impeded Bone Development through Increasing Hif3a Expression in DA2B Mice. PLoS Genet. 2014, 10, e1004589. [Google Scholar] [CrossRef]
- Drevytska, T.; Gonchar, E.; Okhai, I.; Lynnyk, O.; Mankovska, I.; Klionsky, D.; Dosenko, V.; Linnyk, O. The protective effect of Hif3a RNA interference and HIF-prolyl hydroxylase inhibition on cardiomyocytes under anoxia-reoxygenation. Life Sci. 2018, 202, 131–139. [Google Scholar] [CrossRef]
- Bedard, T.; Lowry, R.B. Disease coding systems for arthrogryposis multiplex congenita. Am. J. Med. Genet. Part C Semin. Med. Genet. 2019, 181, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Köhler, S.; Gargano, M.; Matentzoglu, N.; Carmody, L.C.; Lewis-Smith, D.; A Vasilevsky, N.; Danis, D.; Balagura, G.; Baynam, G.; Brower, A.M.; et al. The Human Phenotype Ontology in 2021. Nucleic Acids Res. 2021, 49, D1207–D1217. [Google Scholar] [CrossRef]
- Pergande, M.; Motameny, S.; Özdemir, Ö.; Kreutzer, M.; Wang, H.; Msc, H.-S.D.; Becker, K.; Karakaya, M.; Ehrhardt, H.; Elcioglu, N.; et al. The genomic and clinical landscape of fetal akinesia. Genet. Med. 2020, 22, 511–523. [Google Scholar] [CrossRef]
- Böhm, J.; Malfatti, E.; Oates, E.; Jones, K.; Brochier, G.; Boland, A.; Deleuze, J.-F.; Romero, N.B.; Laporte, J. Novel ASCC1 mutations causing prenatal-onset muscle weakness with arthrogryposis and congenital bone fractures. J. Med. Genet. 2018, 56, 617–621. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.; O’Connell, J.D.; Iocolano, A.D.; Coady, J.A.; Yu, Y.; Gangopadhyay, J.; Gygi, S.P.; Reed, R. The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex. Nucleic Acids Res. 2018, 46, 11939–11951. [Google Scholar] [CrossRef] [PubMed]
- Churchill, L.E.; Delk, P.R.; Wilson, T.E.; Torres-Martinez, W.; Rouse, C.E.; Marine, M.B.; Piechan, J.L. Fetal MRI and ultrasound findings of a confirmed asparagine synthetase deficiency case. Prenat. Diagn. 2020, 40, 1343–1347. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, F.P.; Curry, C.J.; Hevner, R.; Elliott, S.; Fisher, J.H.; Turocy, J.; Dobyns, W.B.; Costa, L.A.; Freitas, E.; Kitajima, J.P.; et al. Biallelic loss of function variants in ATP1A2 cause hydrops fetalis, microcephaly, arthrogryposis and extensive cortical malformations. Eur. J. Med. Genet. 2020, 63, 103624. [Google Scholar] [CrossRef]
- Mohassel, P.; Liewluck, T.; Hu, Y.; Ezzo, D.; Ogata, T.; Saade, D.; Neuhaus, S.; Bolduc, V.; Zou, Y.; Donkervoort, S.; et al. Dominant collagen XII mutations cause a distal myopathy. Ann. Clin. Transl. Neurol. 2019, 6, 1980–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Cerdá, C.; Girós, M.L.; Serrano, M.; Ecay, M.J.; Gort, L.; Dueñas, B.P.; Medrano, C.; García-Alix, A.; Artuch, R.; Briones, P.; et al. A Population-Based Study on Congenital Disorders of Protein N- and Combined with O-Glycosylation Experience in Clinical and Genetic Diagnosis. J. Pediatr. 2017, 183, 170–177. [Google Scholar] [CrossRef]
- Lieu, M.T.; Ng, B.G.; Rush, J.S.; Wood, T.; Basehore, M.J.; Hegde, M.; Chang, R.C.; Abdenur, J.E.; Freeze, H.H.; Wang, R.Y. Severe, fatal multisystem manifestations in a patient with dolichol kinase-congenital disorder of glycosylation. Mol. Genet. Metab. 2013, 110, 484–489. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, N.; Kumaki, T.; Murakami, H.; Enomoto, Y.; Katsumata, K.; Toyoshima, K.; Kurosawa, K. Arthrogryposis multiplex congenita with polymicrogyria and infantile encephalopathy caused by a novel GRIN1 variant. Hum. Genome Var. 2020, 7, 1–4. [Google Scholar] [CrossRef]
- Chen, K.; Yang, K.; Luo, S.; Chen, C.; Wang, Y.; Wang, Y.; Li, D.; Yang, Y.; Tang, Y.; Liu, F.; et al. A homozygous missense variant in HSD17B4 identified in a consanguineous Chinese Han family with type II Perrault syndrome. BMC Med. Genet. 2017, 18, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Itaka, K.; Baba, M.; Uchida, S.; Ishii, T.; Kataoka, K. Muscle-targeted hydrodynamic gene introduction of insulin-like growth factor-1 using polyplex nanomicelle to treat peripheral nerve injury. J. Control. Release 2014, 183, 27–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeffries, L.; Olivieri, J.E.; Ji, W.; Spencer-Manzon, M.; Bale, A.; Konstantino, M.; Lakhani, S.A. Two siblings with a novel nonsense variant provide further delineation of the spectrum of recessive KLHL7 diseases. Eur. J. Med. Genet. 2019, 62, 103551. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.X.; Talbot, J.C.; Teets, E.M.; Previs, S.; Martin, B.L.; Shively, K.M.; Marvin, C.T.; Aylsworth, A.S.; Saadeh-Haddad, R.; Schatz, U.A.; et al. Mutations in MYLPF cause a novel segmental amyoplasia that manifests as distal arthrogryposis. Am. J. Hum. Genet. 2020, 107, 293–310. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Bi, B.; Zhao, P.; Cai, X.; Wan, C.; Shao, J.; He, X. Severe congenital microcephaly with 16p13.11 microdeletion combined with NDE1 mutation, a case report and literature review. BMC Med. Genet. 2017, 18, 141. [Google Scholar] [CrossRef] [Green Version]
- Bonnin, E.; Cabochette, P.; Filosa, A.; Jühlen, R.; Komatsuzaki, S.; Hezwani, M.; Dickmanns, A.; Martinelli, V.; Vermeersch, M.; Supply, L.; et al. Biallelic mutations in nucleoporin NUP88 cause lethal fetal akinesia deformation sequence. PLoS Genet. 2018, 14, e1007845. [Google Scholar] [CrossRef] [Green Version]
- Garcia, A.M.G.; Tutmaher, M.S.; Upadhyayula, S.R.; Russo, R.S.; Verma, S. Novel PLEC gene variants causing congenital myasthenic syndrome. Muscle Nerve 2019, 60, E40–E43. [Google Scholar] [CrossRef]
- Abdel-Salam, G.M.H.; Miyake, N.; Abdel-Hamid, M.S.; Sayed, I.S.M.; Gadelhak, M.I.; Ismail, S.I.; Aglan, M.S.; Afifi, H.H.; Temtamy, S.A.; Matsumoto, N. Phenotypic and molecular insights into PQBP1 -related intellectual disability. Am. J. Med. Genet. Part A 2018, 176, 2446–2450. [Google Scholar] [CrossRef]
- Genini, S.; Nguyen, T.T.; Malek, M.; Talbot, R.; Gebert, S.; Rohrer, G.; Nonneman, D.; Stranzinger, G.; Vögeli, P. Radiation hybrid mapping of 18 positional and physiological candidate genes for arthrogryposis multiplex congenita on porcine chromosome 5. Anim. Genet. 2006, 37, 239–244. [Google Scholar] [CrossRef]
- Gardella, E.; Møller, R. Phenotypic and genetic spectrum of SCN 8A -related disorders, treatment options, and outcomes. Epilepsia 2019, 60, S77–S85. [Google Scholar] [CrossRef] [Green Version]
- Seidahmed, M.Z.; Al-Kindi, A.; Alsaif, H.S.; Miqdad, A.; Alabbad, N.; Alfifi, A.; Abdelbasit, O.B.; Alhussein, K.; Alsamadi, A.; Ibrahim, N.; et al. Recessive mutations in SCYL2 cause a novel syndromic form of arthrogryposis in humans. Qual. Life Res. 2020, 139, 513–519. [Google Scholar] [CrossRef]
- Hakonen, A.H.; Polvi, A.; Saloranta, C.; Paetau, A.; Heikkilä, P.; Almusa, H.; Ellonen, P.; Jakkula, E.; Saarela, J.; Aittomäki, K. SLC18A3 variants lead to fetal akinesia deformation sequence early in pregnancy. Am. J. Med. Genet. Part A 2019, 179, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Magini, P.; Smits, D.J.; Vandervore, L.; Schot, R.; Columbaro, M.; Kasteleijn, E.; van der Ent, M.; Palombo, F.; Lequin, M.H.; Dremmen, M.; et al. Loss of SMPD4 Causes a Developmental Disorder Characterized by Microcephaly and Congenital Arthrogryposis. Am. J. Hum. Genet. 2019, 105, 689–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ravenscroft, G.; Clayton, J.S.; Faiz, F.; Sivadorai, P.; Milnes, D.; Cincotta, R.; Moon, P.; Kamien, B.; Edwards, M.; Delatycki, M.; et al. Neurogenetic fetal akinesia and arthrogryposis: Genetics, expanding genotype-phenotypes and functional genomics. J. Med. Genet. 2020, 15, 106901. [Google Scholar] [CrossRef]
- Montes-Chinea, N.I.; Guan, Z.; Coutts, M.; Vidal, C.; Courel, S.; Rebelo, A.P.; Abreu, L.; Zuchner, S.; Littleton, J.T.; Saporta, M.A. Identification of a new SYT2 variant validates an unusual distal motor neuropathy phenotype. Neurol. Genet. 2018, 4, e282. [Google Scholar] [CrossRef] [Green Version]
- Loeys, B.; Chen, J.; Neptune, E.R.; Judge, D.; Podowski, M.; Holm, T.; Meyers, J.; Leitch, C.C.; Katsanis, N.; Sharifi, N.; et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat. Genet. 2005, 37, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Woolnough, R.; Dhawan, A.; Dow, K.; Walia, J.S. Are Patients with Loeys-Dietz Syndrome Misdiagnosed with Beals Syndrome? Pediatrics 2017, 139, 139–e20161281. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Homeostasis: mechanisms include genes associated with early-onset nuclear DNA excision/repair disorders (ERCC1, ERCC2, and ERCC6) [13]. Monoallelic mutations in ERCC1 are associated with cerebro-oculo-facio-skeletal syndrome. ERLIN1 encodes for a lipid raft-associated protein localized to the mitochondrion and nuclear envelope, and is a component of the ERLIN1/ERLIN2 complex. The complex mediates the endoplasmic reticulum-associated degradation of inositol 1,4,5-trisphosphate receptors (ITPRs) which are important in calcium homeostasis [14]. BAG3, BIN1, ERCC1, ERCC2, ERCC6, ERLIN2, SELENON |
| Cytoskeleton: matrix proteins involved in the sarcomere such as nebulin, a giant protein of thick and thin filaments of striated muscle, encoded by NEB. Mutations in NEB are responsible for the majority of cases of nemaline myopathy [15] which can be diagnosed by Gomori trichrome staining on a muscle biopsy or by electron microscopic preparation. ACTA1, a member of the cytoskeletal grouping, encodes the principal skeletal muscle isoform of adult skeletal muscle, alpha-actin. Residing in the core of the thin filament of the sarcomere, it assists in the generation of muscle contraction [16] ACTA1, ACTB, ACTG1, AP1S2, COL12A1, COL13A1, COL1A1, COL1A2, COL2A1, COL3A1, COL6A1, COL6A2, COL6A3, COLEC11, DCX, DES, DYNC1H1, EMD, FBN1, FBN2, FLNA, FLNB, HSPG2, LMNA, NEB, SPTBN4, SYNE1, TBCD |
| Extra Cellular Matrix: Extracellular matrix (ECM) protein-associated genes include ADAMTS10 and DCHS1. ADAMTS10 is a zinc-dependent protease composed of one cysteine-rich domain, and five thrombospondin type 1 (THBS1) repeats and plays an important role in the formation of the extracellular matrix [17]. DHCS1 is a member of the protocadherin superfamily and encodes a transmembrane cell adhesion molecule responsible for apical anchoring in the brain [18]. ADAMTS10, CDON, DCHS1, MMP2, RAPSN |
| Signal Transduction: Promotes signaling within a cell via enzyme network cascades to generate precise and appropriate physiologic responses, particularly in skeletal development. FGFR3 codes for an important tyrosine kinase signal transducer in chondrocytes, functioning to attenuate cartilage growth. FGFR 1–4 transmit at least 18 different fibroblast growth factor (FGF) ligands, therefore, exhibiting a variety of physiological functions [19]. GDF5 fulfills important functions with respect to bone and muscle [20]. Through its high affinity for BMPR1B, GDF5 positively regulates chondrogenesis, leading to SMAD signal transduction [21]. Through NOG mediated interaction, GDF5 paradoxically also negatively regulates chondrogenesis. ADGRG6, CAVIN1, CCDC22, CD96, CFL2, CRLF1, CRTAP, DOK7, EBP, FGFR1, FGFR2, FGFR3, GDF5, IFIH1, KIAA0586, MAGEL2, NF1, PEX5, PEX7, PMP22, RAB3GAP1, RAB3GAP2, STAC3, WNT5A, KBTBD13 |
| Proto-oncogenes: Proto-oncogenes act to facilitate dysregulated cell growth and differentiation. Mutations in HRAS are associated with Costello syndrome, characterized by distinct facial features, papilloma of the face, cardiac anomalies, growth restriction, developmental delays, and tumor predisposition. An HRAS mutation was identified in an infant with features of Costello syndrome and distal arthrogryposis [22]. AKT1, CBL, HRAS, RAB18, RET, SKI |
| Enzyme: Account for the largest category of genes identified through IPA analysis. 7-dehydrocholesterol reductase (DHCR7) encodes the penultimate step in the cholesterol biosynthetic pathway. Smith-Lemli-Opitz Syndrome is an autosomal recessive disorder caused by an inherited deficiency of DHCR7 which is associated with a variety of birth defects, joint contractures, and intellectual disability [23]. UBE3 which encodes E3 ubiquitin-protein ligase, a maternally expressed imprinted E3 ubiquitin-protein ligase expressed mainly in the brain, is an integral part of the ubiquitin protein degradation system. Angelman syndrome, characterized by severe cognitive impairment, seizures, an ataxic puppet-like gait, and paroxysms of laughter, is caused by an absence of expression of maternal UBE3A [24]. ALG2, ASAH1, B3GAT3, CANT1, CHAT, CHST14, CHST3, DHCR7, DPAGT1, DSE, ECEL1, EXTL3, EZH2, FBXL4, FKRP, FUCA1, GAD1, GBA, GFPT1, GUSB, HSD17B4, INPP5K, LARGE1, MASP1, MTM1, NAA10, NEU1, OCRL, P3H1, PAFAH1B1, PHGDH, PLOD1, PLOD2, PLOD3, PMM2, POLR3A, POMT1, POMT2, POR, PPIB, PPP3CA, PSAT1, PTDSS1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, TOR1A, TREX1, UBA1, UBE3A, ZMPSTE24 |
| Transcription Factor/regulation: Transcription factors have a pivotal role in the regulation of genes associated with limb and muscle development. T-Box Transcription Factor 5 (TBX5) mutations are associated with Holt Oram syndrome characterized by upper limb defects and cardiac malformations [25,26]. TRIP4 encodes ASC-1, a transcription co-activator. Infants with TRIP4 mutations present with a congenital muscular dystrophy and respiratory failure. Muscle biopsy shows decreased mitochondria and sarcomere disorganization [27]. ARX, ASXL3, ATN1, ATRX, AUTS2, EGR2, FGD1, GZF1, IGHMBP2, IRF6, LMX1B, MED12, NSD1, PAX3, PLEKHG5, PQBP1, RBM10, SETBP1, SETX, SHOX, SOX9, TBX5, TRIP4, ZC4H2, ZEB2, ZIC2 |
| Mitochondria: Mitochondria are depended upon highly by the brain and skeletal muscle tissues for energy. Ganglioside Differentiation Associated Protein 1 (GDAP1) encodes a mitochondrial protein postulated to play a role in signal transduction in the brain. Mutations in GDAP1 are associated with various subtypes of the hereditary and sensory-motor neuropathy disease Charcot Marie Tooth (CMT), including an autosomal recessive intermediate type [28,29,30,31,32]. RMRP codes for non-coding RNA involved in mitochondrial DNA replication through the encoding of a mitochondrial RNA processing endonuclease which cleaves mitochondrial RNA at a priming site necessary for mitochondrial DNA replication. Mutations in RMRP are associated with cartilage-hair hypoplasia [33]. RMRP is essential for early murine development [34]. ATAD3A, C12orf65, GDAP1, GFM2, MFN2, RMRP, SPAR |
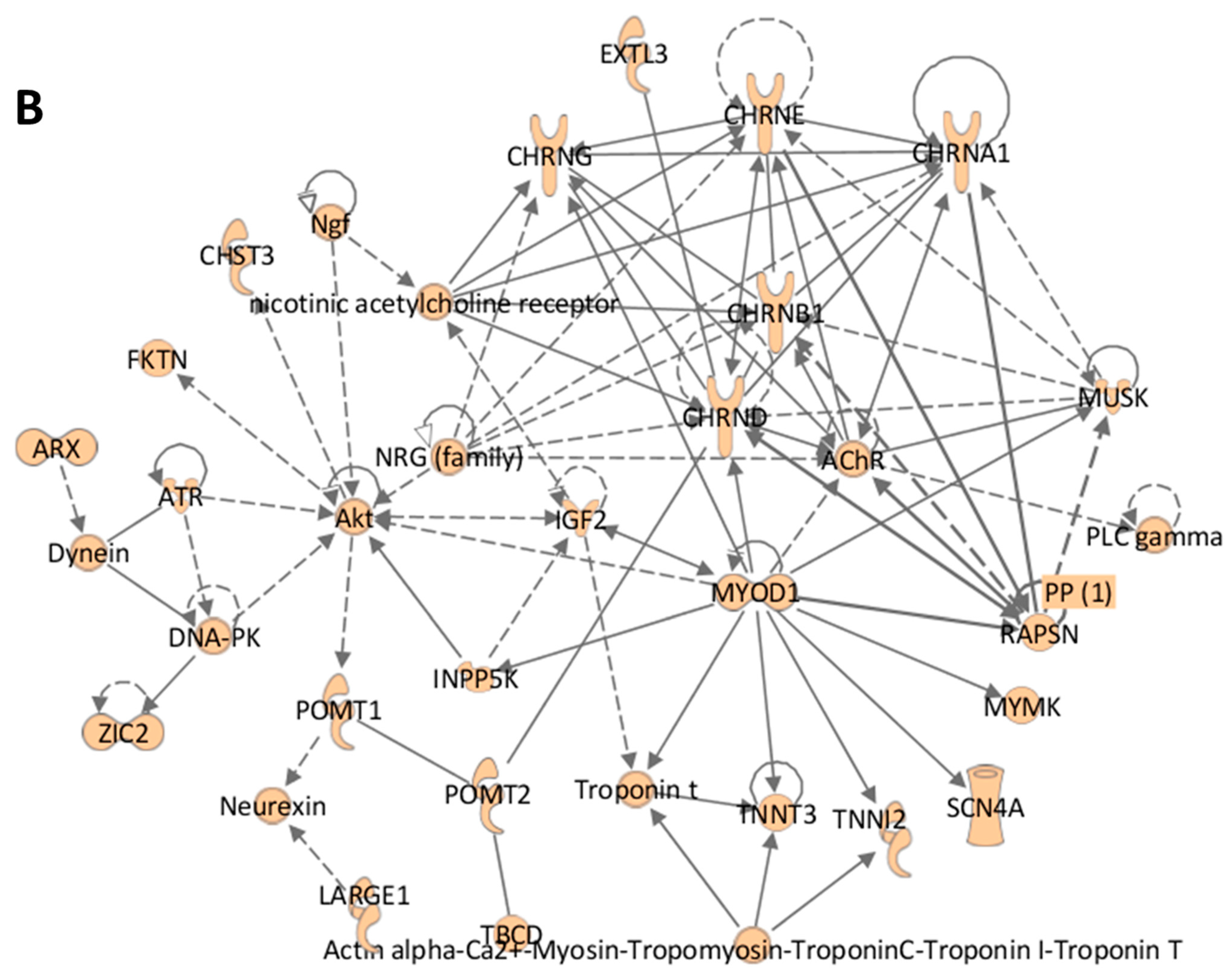
| Membrane Receptor/Ion Channel: Membrane receptor and ion channels is the second largest group of affected genes leading to AMC-SC. CHRNA1 (cholinergic receptor nicotinic receptor alpha 1 subunit 1) is one of 5 subunits of the acetylcholine receptor (AChR). This gene encodes an alpha subunit and functions as part of acetylcholine binding and channel. Mutations in CHRNA1 are associated with lethal multiple pterygium syndrome, characterized by the presence of multiple pterygia, intrauterine growth retardation, and flexion contractures resulting in severe arthrogryposis and fetal akinesia [35]. PIEZO2 is postulated to function as an integral part of mechanically activated cation channel in somatosensory neurons through establishing connections between mechanical forces and biological signals. Mutations in PIEZO2 are associated with distal arthrogryposis type 5, Gordon syndrome, and Marden–Walker syndrome [36]. ATP7A, CHRNA1, CHRNB1, CHRND, CHRNE, CHRNG, GPC3, GRIN1, KCNA1, KCNH1, MEGF10, NALCN, NRXN1, NUP88, PIEZO2, PIGS, PIGT, ROR2, RYR1, SCN4A, SGCG, SLC12A6, SLC18A3, SLC26A2, SLC2A10, SLC35A3, SLC39A13, SLC5A7, SNAP25, SYT2, TGFBR1, TGFBR2, TRPV4, VAMP1, WASHC5 |
| Kinase: Kinases phosphorylate target molecules for activation or inactivation. ATR encodes a serine/threonine kinase and halts cell cycling entry upon DNA stress to enable DNA repair [37]. Compound heterozygous mutations in ATR are associated with Seckel syndrome characterized by dwarfism, microcephaly, and cognitive impairment [38]. MAP3K7 mediates cellular transduction in response to environmental changes through association with interleukin receptor (ILR1). Through the cytokine IL-1 mediated interaction with the hypothalamic IL-1 receptor, the hypothalamo-pituitary-adrenocortical axis and sympathetic nervous system pathways suppressing bone formation are activated [39]. Fronto-metaphyseal dysplasia, a progressive sclerosing skeletal dysplasia characterized by small bone undermodeling, supraorbital hyperostosis, large and small joint contractures as well as developmental abnormalities, of the cardiorespiratory system and the genitourinary tract is associated with MAP3K7 mutations [40]. ATR, CASK, MAP3K7, MUSK, NEK9, PRKAR1A |
| Intracellular transport: Intracellular transport proteins are structural proteins that facilitate the movement of vesicles and substances within a cell. BICD2 codes for a structural protein functioning as an intracellular adaptor for the dynein motor complex, linking it to various cargos. Through the stabilization of the interaction between dynein and dynactin, the movement of dynein is facilitated along the microtubule [41]. Mono-allelic mutations in BICD2 cause congenital spinal muscular atrophy [42]. GLE1 is postulated to act as a terminal step in the transport of mature messenger RNA messages from the nucleus to the cytoplasm. Bi-allelic mutations in GLE1 are associated with a lethal congenital contracture syndrome characterized by fetal hydrops, degeneration of anterior horn cells, and congenital contractures [43]. BICD2, DYM, FKBP10, GLE1, KIF1A, VPS53 |
| Structural: Structural proteins provide the framework for a cell or complex of cells. The LAMA2 gene encodes laminin-2 or merosin, a major component of the extrasynaptic membrane of muscle cell basement membrane. Laminin-211 binds to the glycosylated residues of alpha-dystroglycan (DAG1) in skeletal muscle fibers [44]. Bi-allelic mutations in LAMA2 are associated merosin-deficient congenital muscular dystrophy. Affected patients have hypotonia, joint contractures and may develop scoliosis. Myosin, the major contractile protein in muscle, is composed of two heavy chains and two light chains. MYH3 encodes the embryonic myosin heavy chain 3. MYH3 mutations appear to reside near a groove that is part of the myosin head and are associated with distal arthrogryposis type 1 in which contractures are limited to distal joints, Freeman –Sheldon, Sheldon -Hall syndromes [45]. Affected patients with Freeman Sheldon and Sheldon Hall syndromes have distal joint contractures, characteristic facial features and may develop scoliosis. MYH3 mutations are postulated to cause structural changes in myosin that potentially alter myosin domain-domain interactions during ATP catalysis or affect nucleotide-binding site conformation. FHL1, FKTN, KLHL41, LAMA2, LMOD3, MYBPC1, MYH2, MYH3, MYMK, MYO18B, MYO9A, MYOD1, MYPN, PRX, TNNI2, TNNT3, TPM2, TPM3, TTN, VMA21 |
| Title | Title | Altered Genes | ||
|---|---|---|---|---|
| Phenotype | Gene 1 | Gene 2 | Gene 3 | |
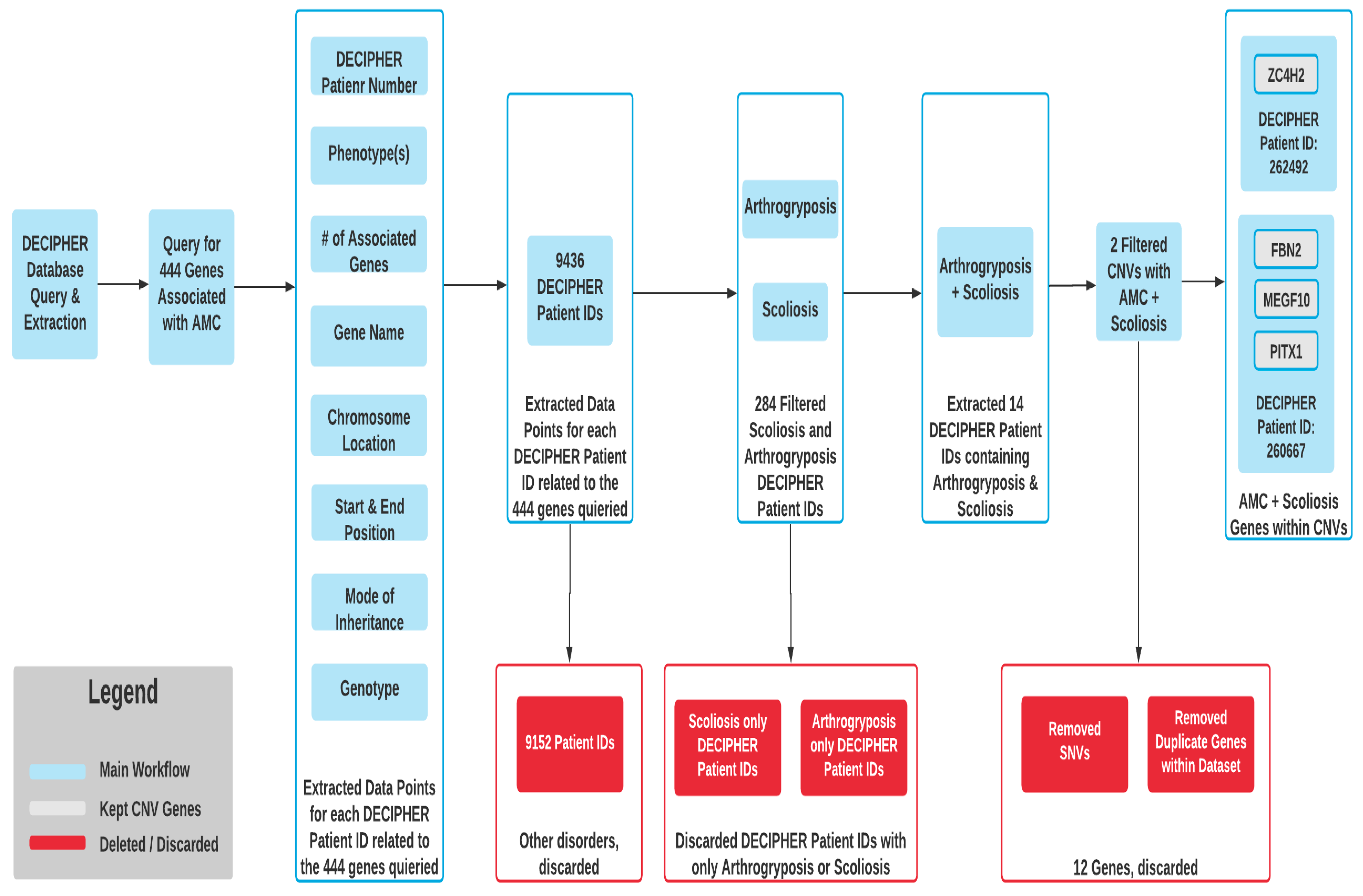
| Patient 1 | Cleft palate, crumpled ear, distal arthrogryposis, intellectual disability, micrognathia, scoliosis, syringomyelia; mild pulmonary stenosis | FBN2 (Chr5, de novo, loss, heterozygous) * | MEGF10 (Ch5, de novo, loss, heterozygous) * | PITX1 (Ch5, de novo, loss, heterozygous) * |
| Patient 2 | AMC, dysphagia, dystonia, global developmental delay, laryngomalacia, thoracolumbar scoliosis | ZC4H2 (ChX, de novo, loss, het) * | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Latypova, X.; Creadore, S.G.; Dahan-Oliel, N.; Gustafson, A.G.; Wei-Hung Hwang, S.; Bedard, T.; Shazand, K.; van Bosse, H.J.P.; Giampietro, P.F.; Dieterich, K. A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita. Genes 2021, 12, 1052. https://doi.org/10.3390/genes12071052
Latypova X, Creadore SG, Dahan-Oliel N, Gustafson AG, Wei-Hung Hwang S, Bedard T, Shazand K, van Bosse HJP, Giampietro PF, Dieterich K. A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita. Genes. 2021; 12(7):1052. https://doi.org/10.3390/genes12071052
Chicago/Turabian StyleLatypova, Xenia, Stefan Giovanni Creadore, Noémi Dahan-Oliel, Anxhela Gjyshi Gustafson, Steven Wei-Hung Hwang, Tanya Bedard, Kamran Shazand, Harold J. P. van Bosse, Philip F. Giampietro, and Klaus Dieterich. 2021. "A Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita" Genes 12, no. 7: 1052. https://doi.org/10.3390/genes12071052