
Recapture Lysosomal Enzyme Deficiency via Targeted Gene Disruption in the Human Near-Haploid Cell Line HAP1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
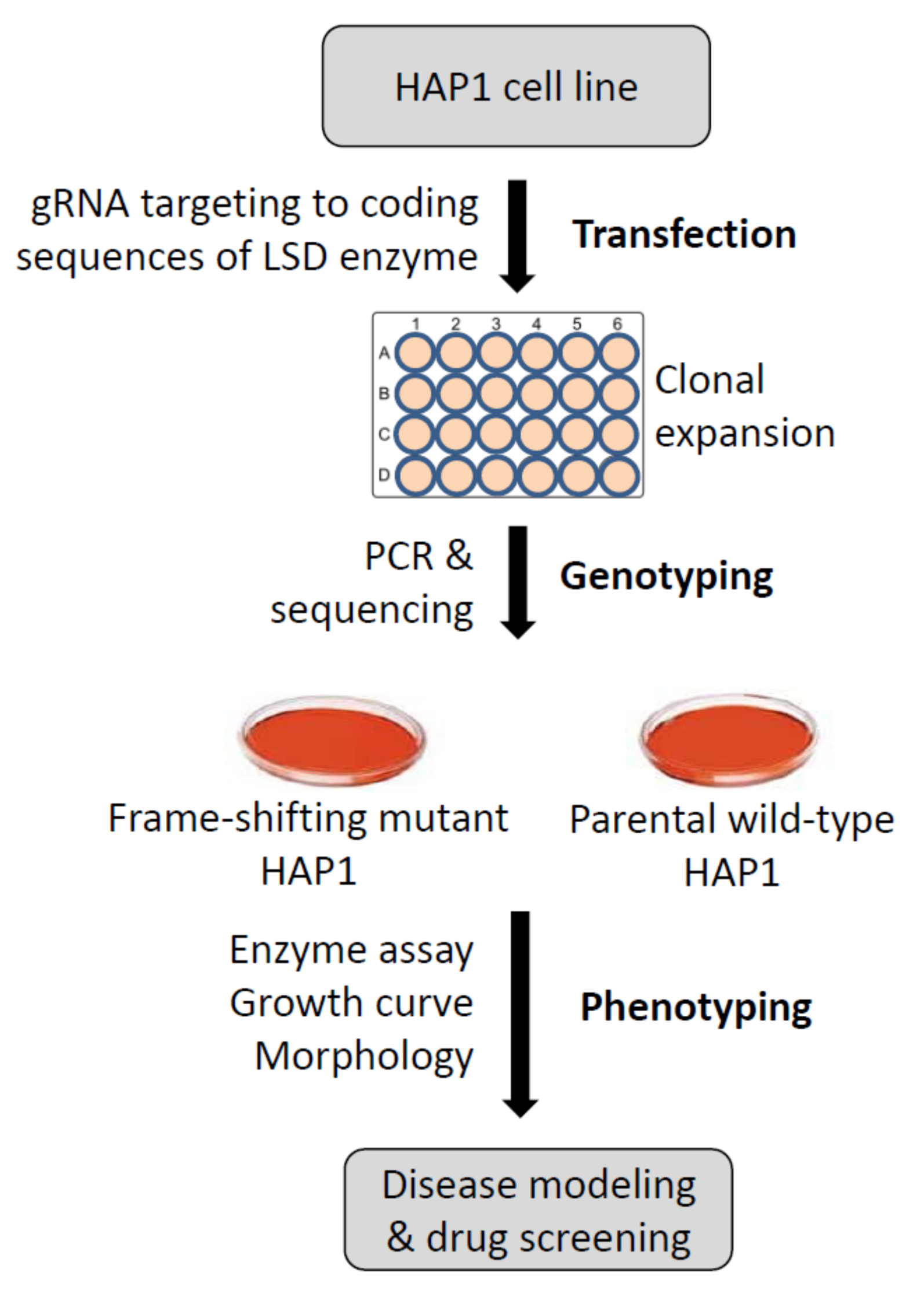
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. GBA Knockout Strategy
2.4. Genomic DNA Extraction and PCR
2.5. GBA Enzyme Assay
2.6. Lysosome Staining
2.7. Fluorescent and Confocal Microscopy
2.8. Data Collection and Statistical Analysis
3. Results
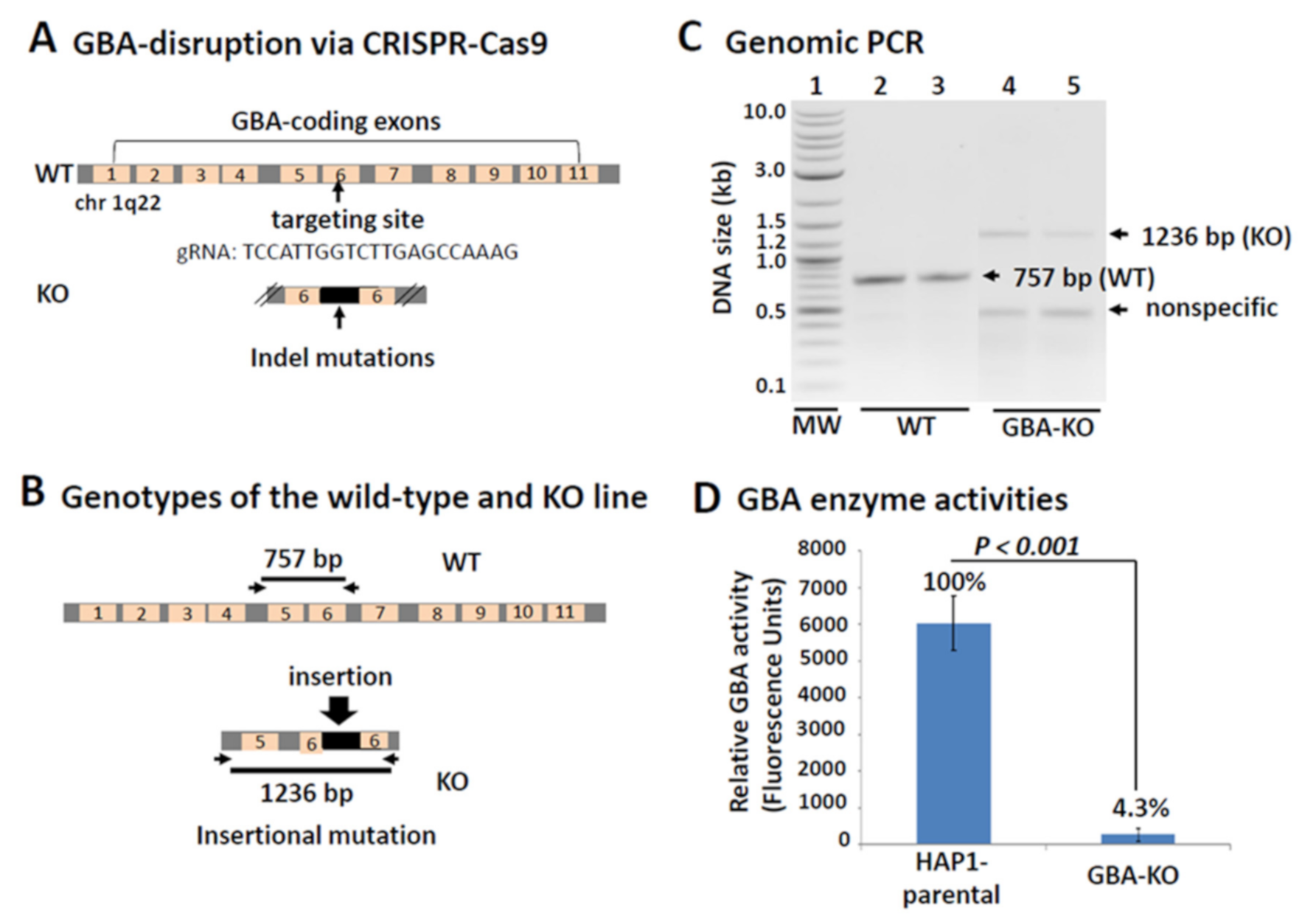
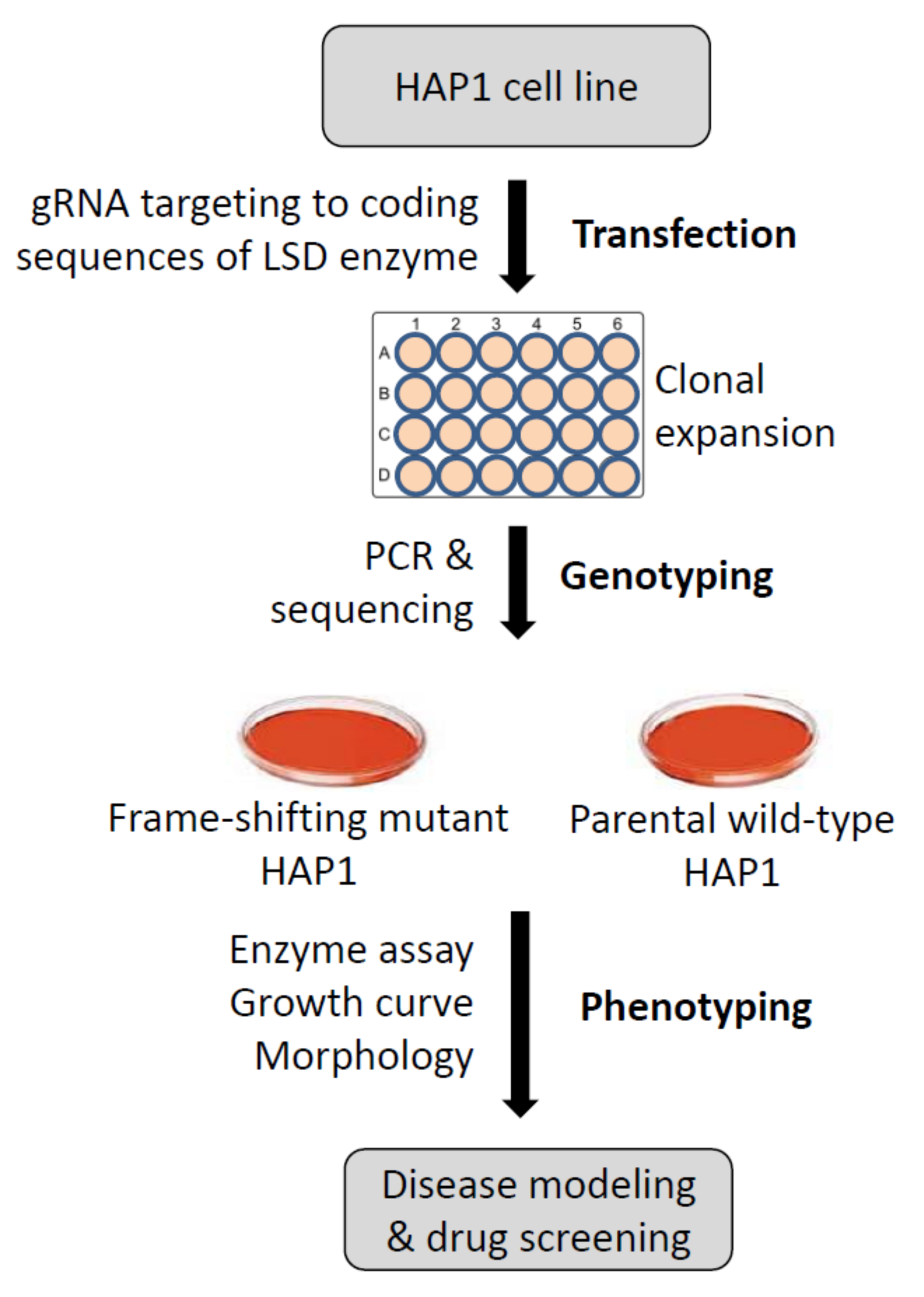
3.1. System Design and Knockout Strategy to Model Human Gaucher Disease
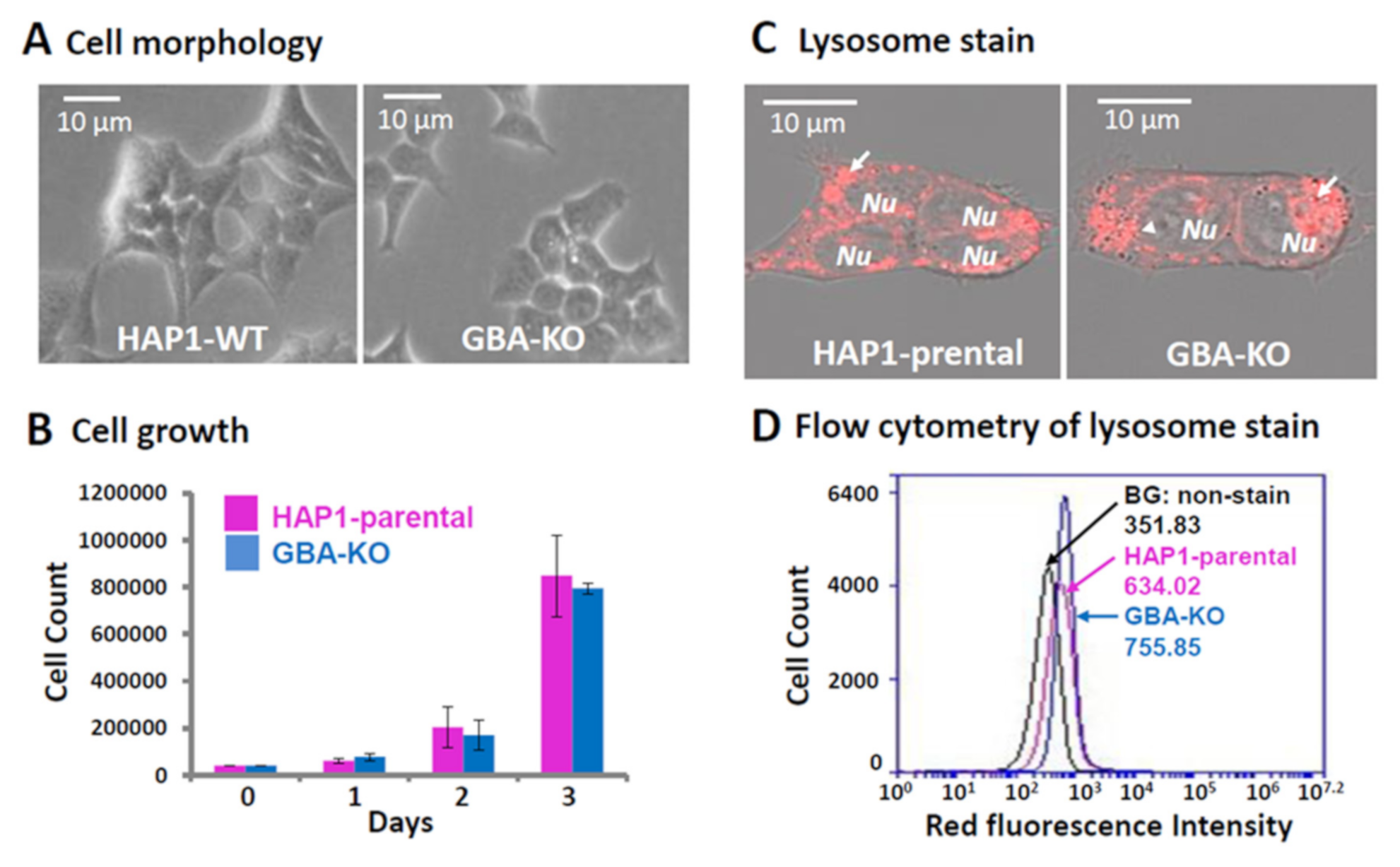
3.2. Characterization of GBA-KO Line
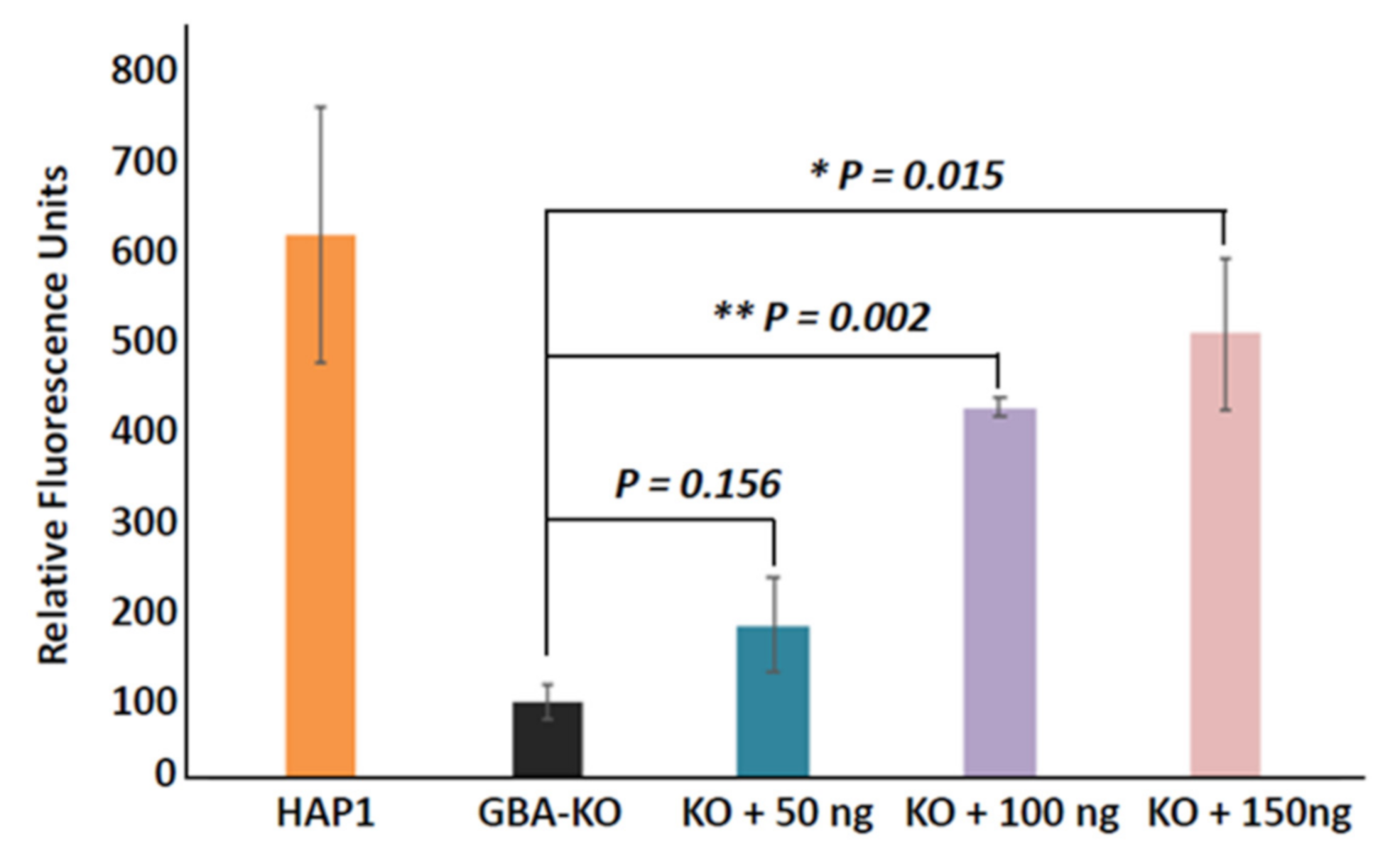
3.3. Enzyme Replacement of GBA in GBA-KO Line by Recombinant Human GBA
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Burrow, T.A.; Hopkin, R.; Leslie, N.D.; Tinkle, B.T.; Grabowski, G.A. Enzyme reconstitution/replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2007, 19, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Bodamer, O.A.; Watson, M.S.; Wilcox, W.R. Lysosomal storage diseases: Diagnostic confirmation and management of presymptomatic individuals. Genet. Med. 2011, 13, 457–484. [Google Scholar] [CrossRef] [Green Version]
- Lachmann, R.H. Enzyme replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2011, 23, 588–593. [Google Scholar] [CrossRef]
- Parenti, G.; Andria, G.; Ballabio, A. Lysosomal storage diseases: From pathophysiology to therapy. Annu. Rev. Med. 2015, 66, 471–486. [Google Scholar] [CrossRef]
- Futerman, A.H.; van Meer, G. The cell biology of lysosomal storage disorders. Nat. Rev. Mol. Cell Biol. 2004, 5, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E. Limitations of enzyme replacement therapy: Current and future. J. Inherit. Metab. Dis. 2006, 29, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Di Bisceglie, A.M.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher’s disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef]
- Farfel-Becker, T.; Vitner, E.B.; Futerman, A.H. Animal models for Gaucher disease research. Dis. Models Mech. 2011, 4, 746–752. [Google Scholar] [CrossRef] [Green Version]
- Enquist, I.B.; Nilsson, E.; Ooka, A.; Månsson, J.-E.; Olsson, K.; Ehinger, M.; Brady, R.O.; Richter, J.; Karlsson, S. Effective cell and gene therapy in a murine model of Gaucher disease. Proc. Natl. Acad. Sci. USA 2006, 103, 13819–13824. [Google Scholar] [CrossRef] [Green Version]
- Hartley, W.J.; Blakemore, W.F. Neurovisceral glucocerebroside storage (Gaucher’s disease) in a dog. Vet. Pathol. 1973, 10, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Koike-Yusa, H.; Li, Y.; Tan, E.-P.; Velasco-Herrera, M.D.C.; Yusa, K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat. Biotechnol. 2014, 32, 267–273. [Google Scholar] [CrossRef]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef]
- Segal, D.J.; Meckler, J.F. Genome engineering at the dawn of the golden age. Annu. Rev. Genom. Hum. Genet. 2013, 14, 135–158. [Google Scholar] [CrossRef]
- Hsu, P.; Lander, E.S.; Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef] [Green Version]
- Uhde-Stone, C.; Sarkar, N.; Antes, T.; Otoc, N.; Kim, Y.; Jiang, Y.J.; Lu, B. A TALEN-based strategy for efficient bi-allelic miRNA ablation in human cells. RNA 2014, 20, 948–955. [Google Scholar] [CrossRef] [Green Version]
- Gaj, T.; Gersbach, C.A.; Barbas, C.F. ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013, 31, 397–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carette, J.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.; Spooner, E.; Ploegh, H.L.; et al. Haploid Genetic Screens in Human Cells Identify Host Factors Used by Pathogens. Science 2009, 326, 1231–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carette, J.E.; Guimaraes, C.P.; Wuethrich, I.; Blomen, V.A.; Varadarajan, M.; Sun, C.; Bell, G.; Yuan, B.; Muellner, M.K.; Nijman, S.M.; et al. Global gene disruption in human cells to assign genes to phenotypes by deep sequencing. Nat. Biotechnol. 2011, 29, 542–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hruska, K.S.; Lamarca, M.E.; Scott, C.R.; Sidransky, E. Gaucher disease: Mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum. Mutat. 2008, 29, 567–583. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, G.A. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet 2008, 372, 1263–1271. [Google Scholar] [CrossRef]
- Barton, N.W.; Furbish, F.S.; Murray, G.J.; Garfield, M.; Brady, R.O. Therapeutic response to intravenous infusions of glucocerebrosidase in a patient with Gaucher disease. Proc. Natl. Acad. Sci. USA 1990, 87, 1913–1916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrow, J. Enzyme replacement therapy for Gaucher disease. Expert Opin. Biol. Ther. 2008, 9, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Do, M.A.; Levy, D.; Brown, A.; Marriott, G.; Lu, B. Targeted delivery of lysosomal enzymes to the endocytic compartment in human cells using engineered extracellular vesicles. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchler-Bauer, A.; Bo, Y.; Han, L.; He, J.; Lanczycki, C.J.; Lu, S.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; et al. CDD/SPARCLE: Functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017, 45, D200–D203. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, H.; Tsirigos, K.; Brunak, S.; Von Heijne, G. A Brief History of Protein Sorting Prediction. Protein J. 2019, 38, 200–216. [Google Scholar] [CrossRef] [Green Version]
- Gersbach, C.A. Genome engineering: The next genomic revolution. Nat. Methods 2014, 11, 1009–1011. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, A.; Zhang, J.; Lawler, B.; Lu, B. Recapture Lysosomal Enzyme Deficiency via Targeted Gene Disruption in the Human Near-Haploid Cell Line HAP1. Genes 2021, 12, 1076. https://doi.org/10.3390/genes12071076
Brown A, Zhang J, Lawler B, Lu B. Recapture Lysosomal Enzyme Deficiency via Targeted Gene Disruption in the Human Near-Haploid Cell Line HAP1. Genes. 2021; 12(7):1076. https://doi.org/10.3390/genes12071076
Chicago/Turabian StyleBrown, Annie, Jiayi Zhang, Brendan Lawler, and Biao Lu. 2021. "Recapture Lysosomal Enzyme Deficiency via Targeted Gene Disruption in the Human Near-Haploid Cell Line HAP1" Genes 12, no. 7: 1076. https://doi.org/10.3390/genes12071076
APA StyleBrown, A., Zhang, J., Lawler, B., & Lu, B. (2021). Recapture Lysosomal Enzyme Deficiency via Targeted Gene Disruption in the Human Near-Haploid Cell Line HAP1. Genes, 12(7), 1076. https://doi.org/10.3390/genes12071076