
Clinicopathological and Genomic Profiles of Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma Identify Overlapping Signatures with a High Mutational Burden


 , , ,
, , , 
Abstract
:1. Introduction
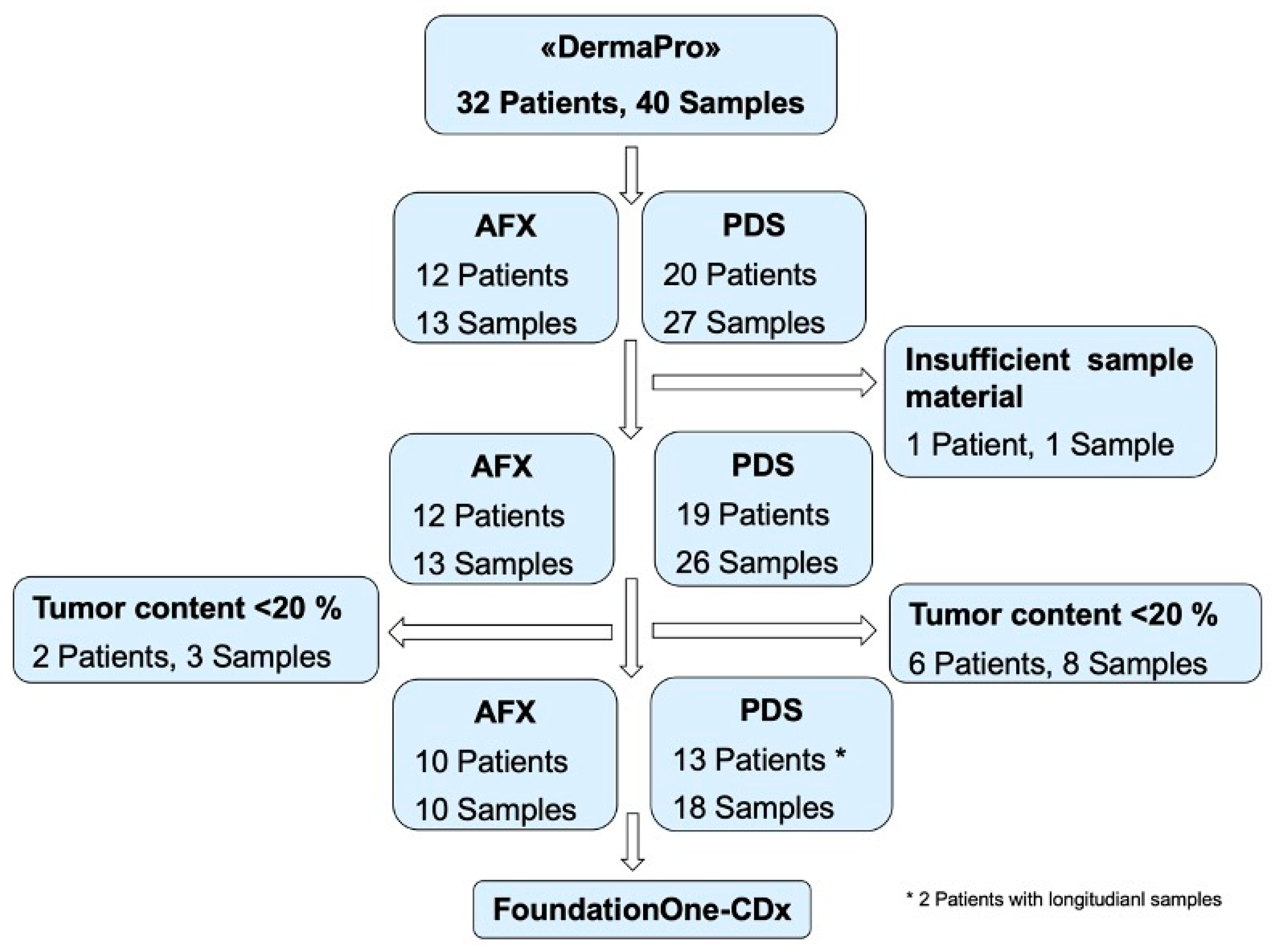
2. Materials and Methods
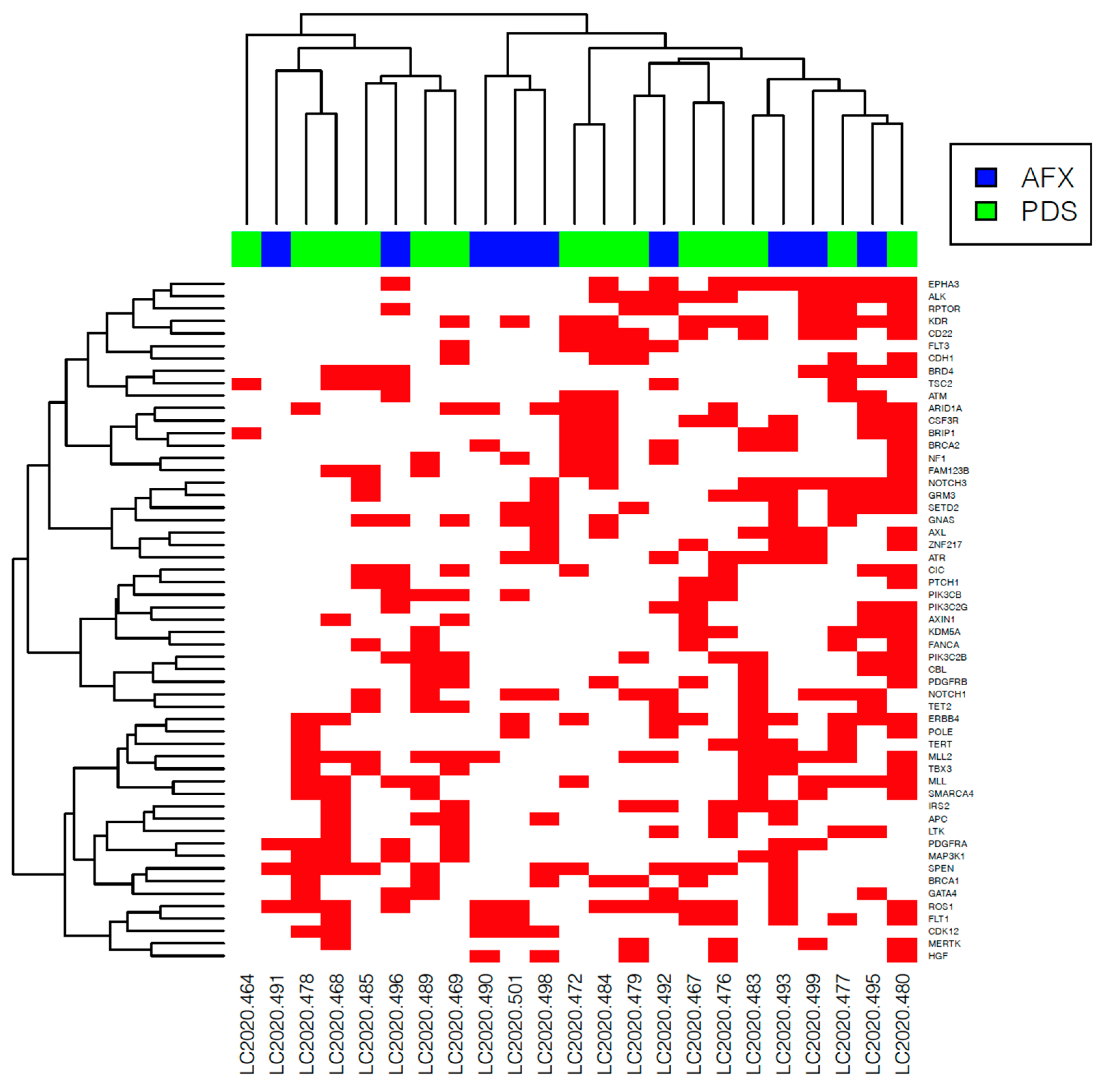
3. Results
3.1. Study Cohort
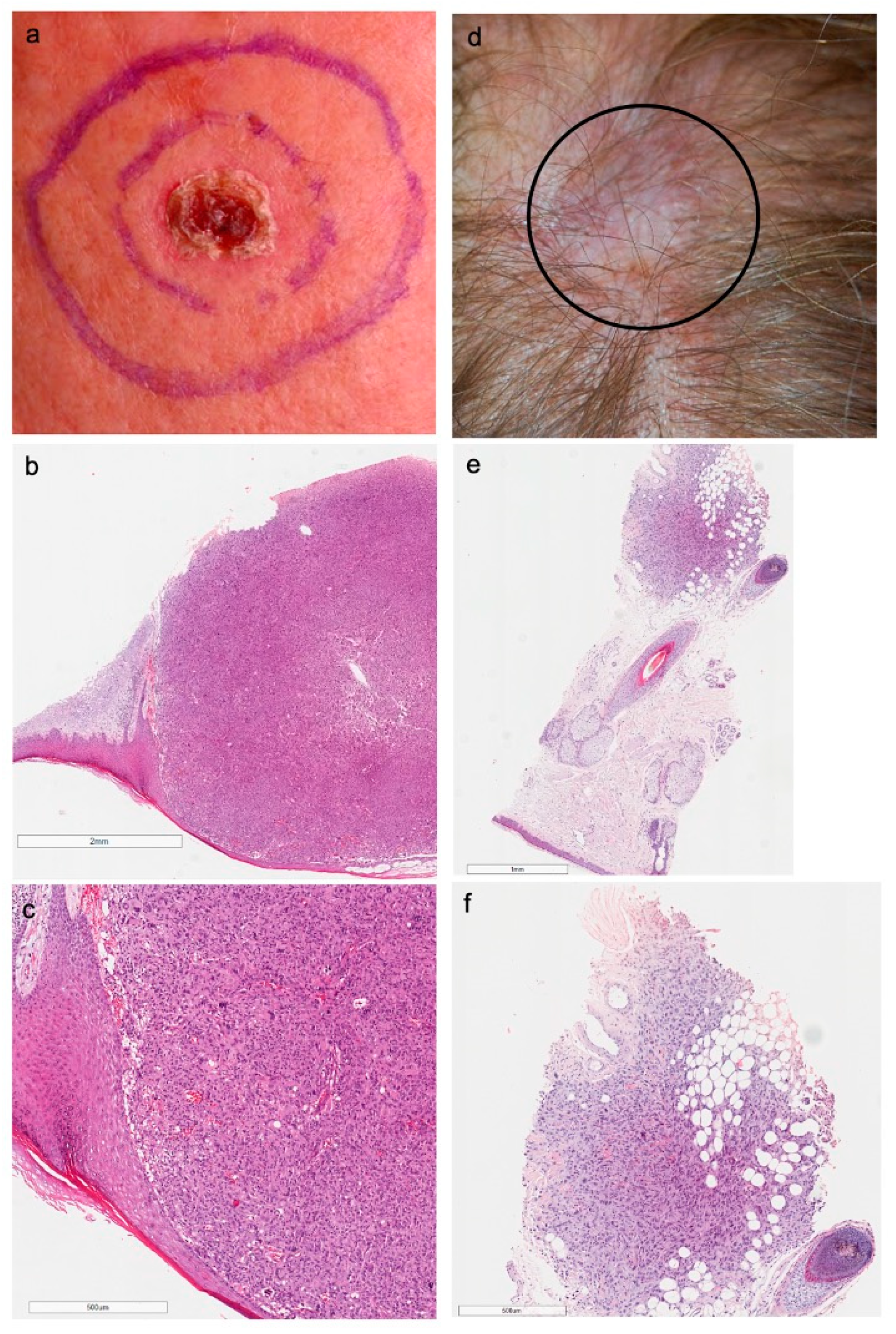
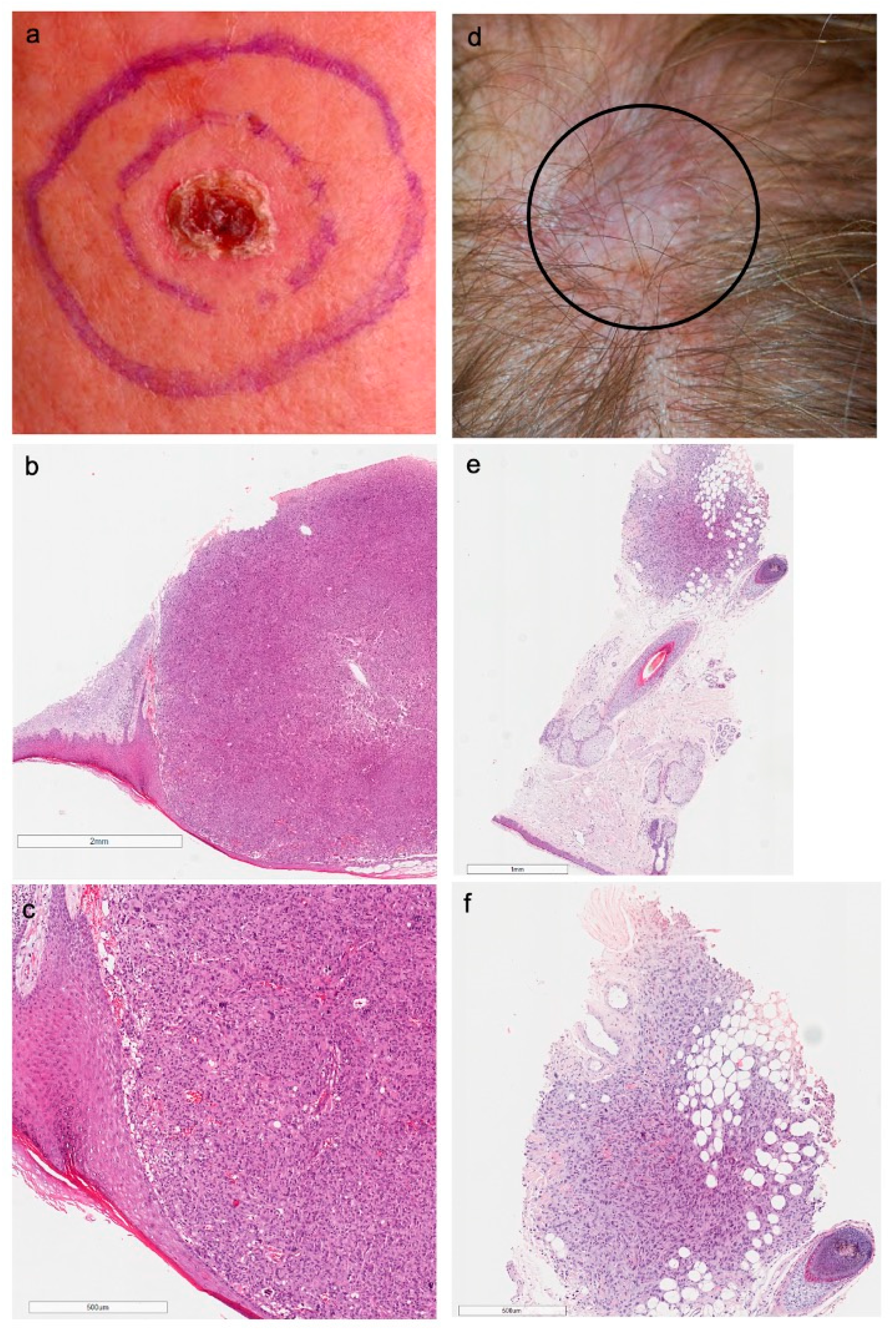
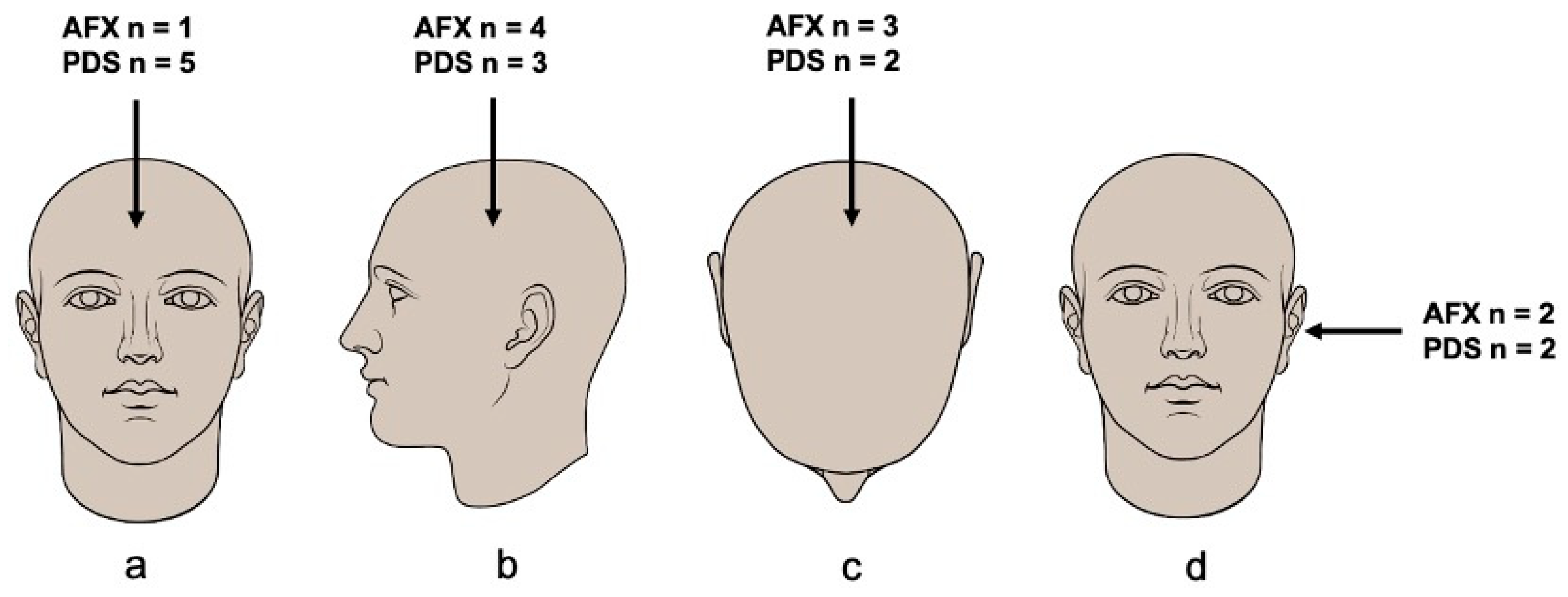
3.2. Clinicopathological Features
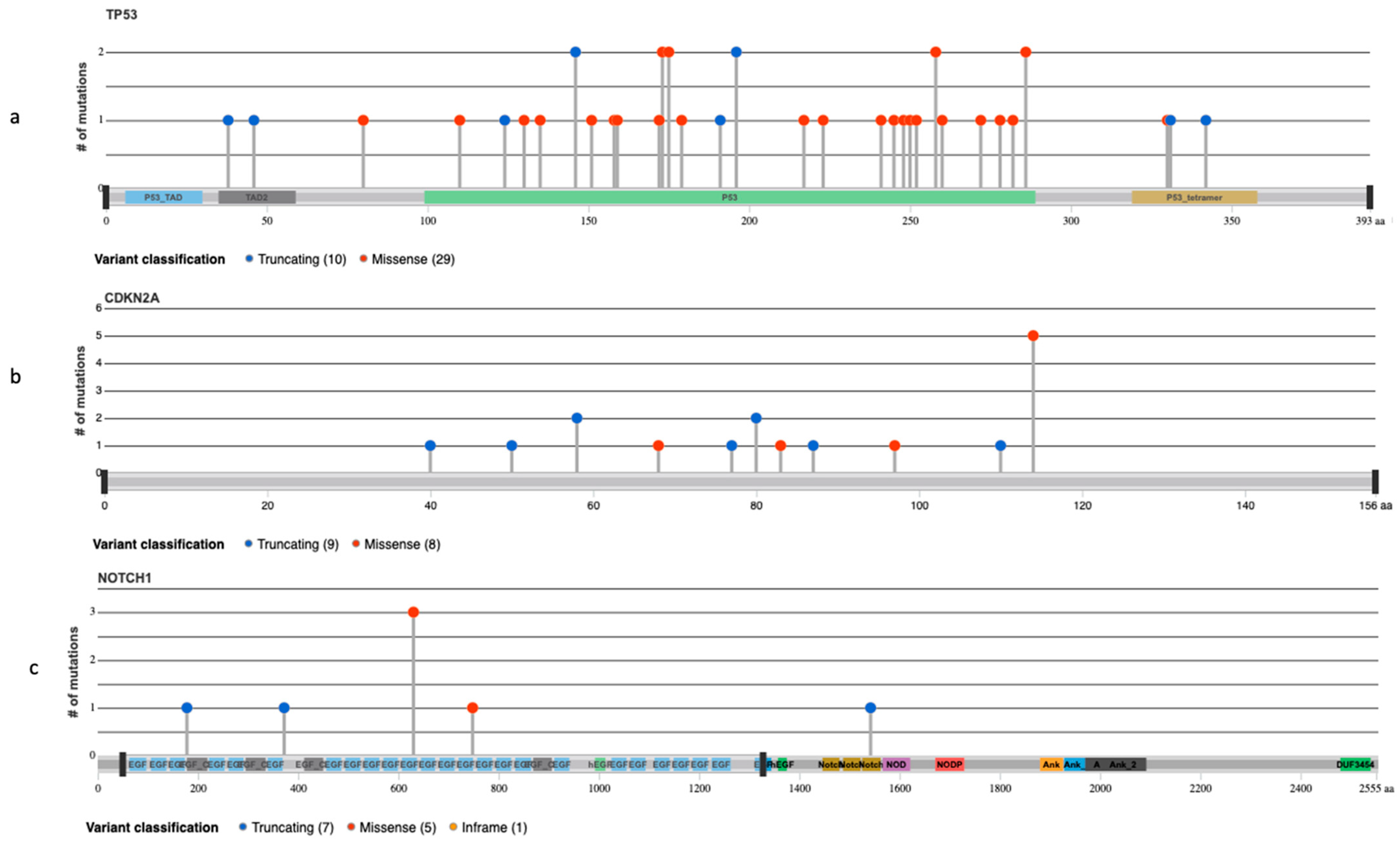
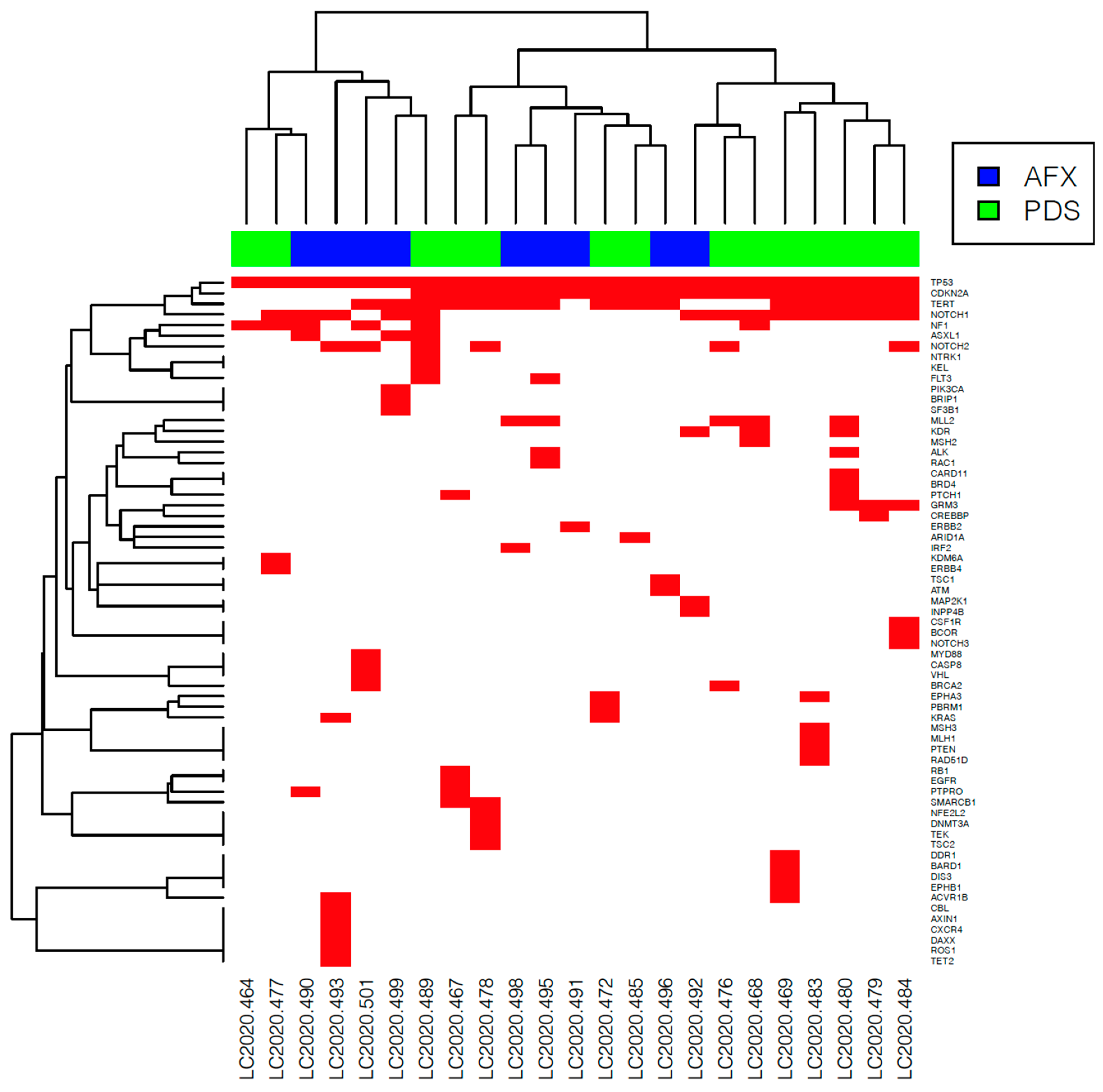
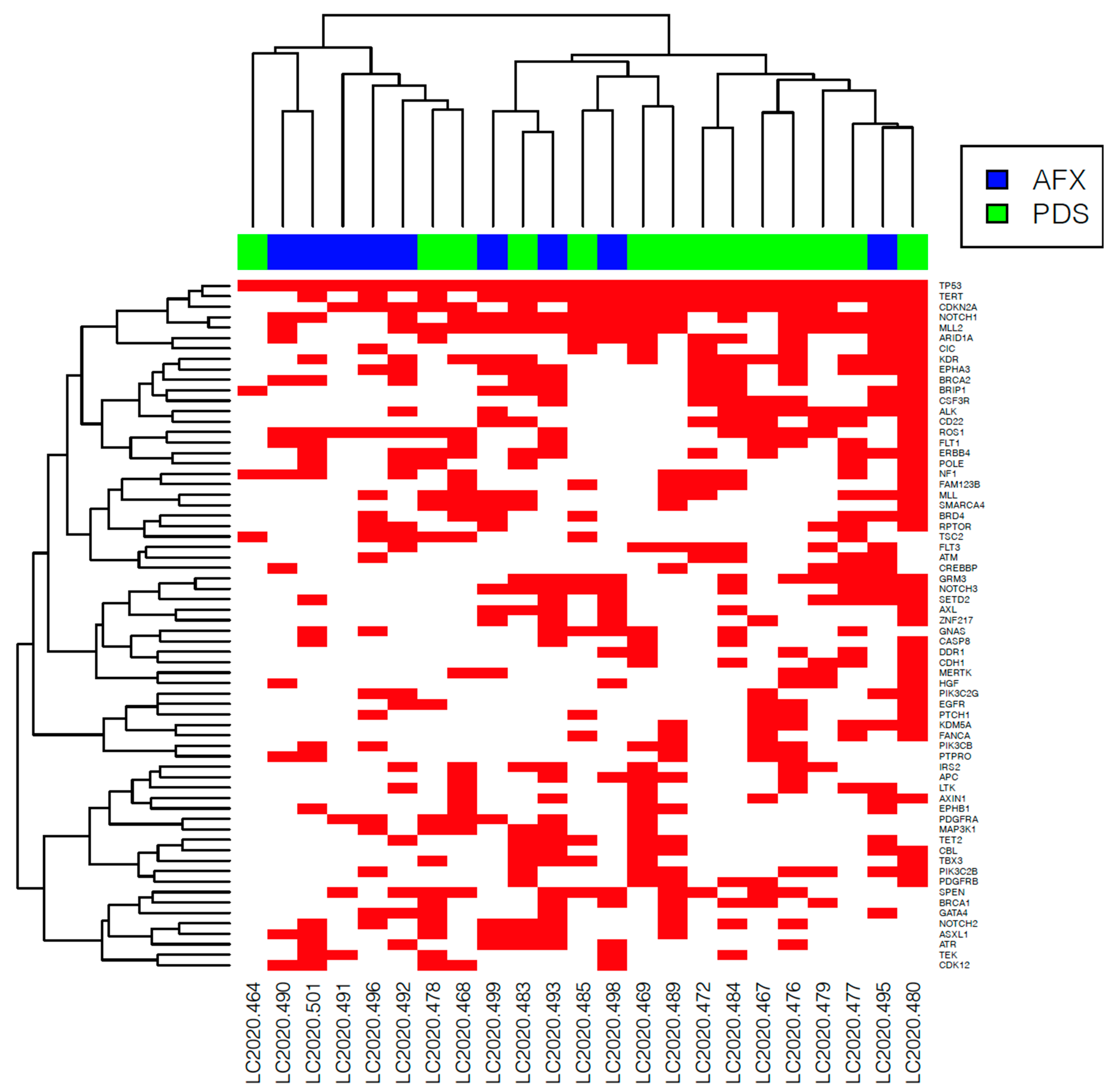
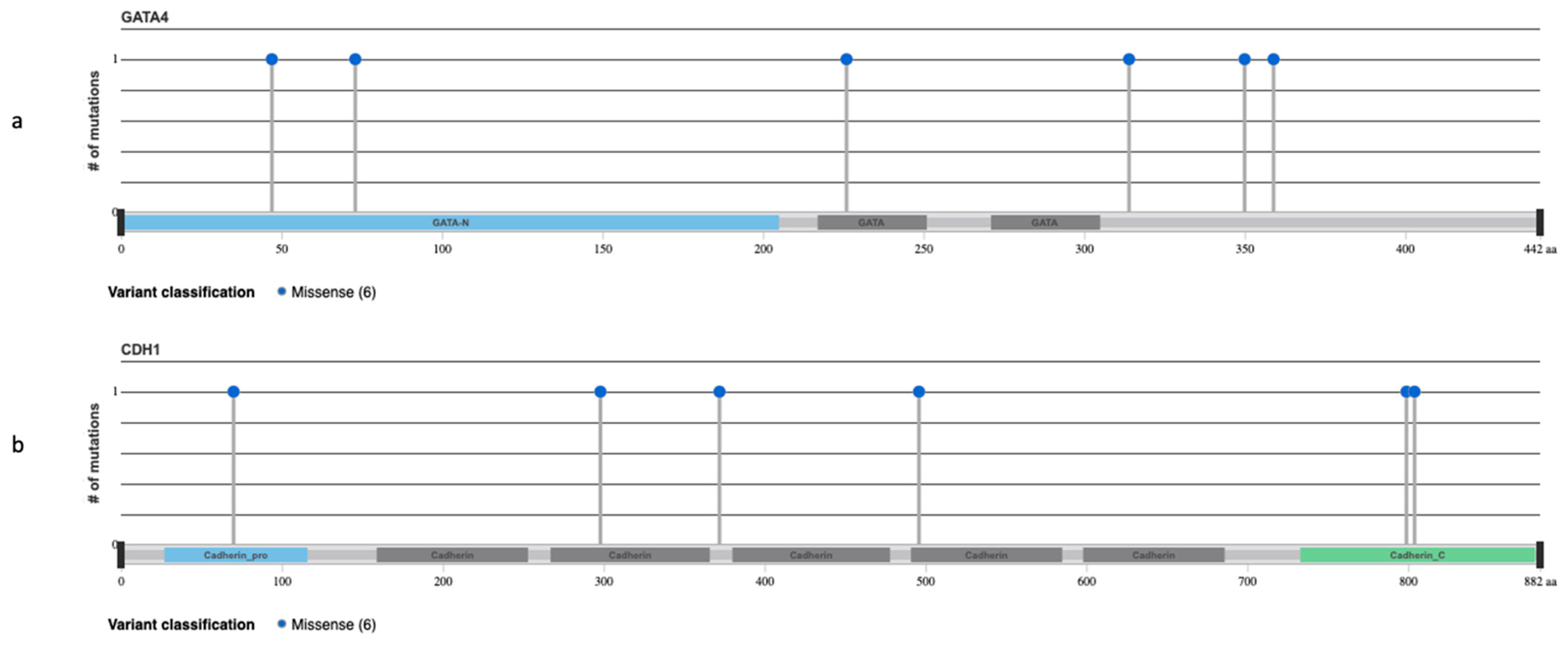
3.3. NGS Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Soleymani, T.; Aasi, S.Z.; Novoa, R.; Hollmig, S.T. Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma: Updates on Classification and Management. Dermatol. Clin. 2019, 37, 253–259. [Google Scholar] [CrossRef]
- Fretzin, D.F.; Helwig, E.B. Atypical fibroxanthoma of the skin. A clinicopathologic study of 140 cases. Cancer 1973, 31, 1541–1552. [Google Scholar] [CrossRef]
- Soleymani, T.; Tyler Hollmig, S. Conception and Management of a Poorly Understood Spectrum of Dermatologic Neoplasms: Atypical Fibroxanthoma, Pleomorphic Dermal Sarcoma, and Undifferentiated Pleomorphic Sarcoma. Curr. Treat. Options Oncol. 2017, 18, 50. [Google Scholar] [CrossRef]
- Koch, M.; Freundl, A.J.; Agaimy, A.; Kiesewetter, F.; Kunzel, J.; Cicha, I.; Alexiou, C. Atypical Fibroxanthoma–Histological Diagnosis, Immunohistochemical Markers and Concepts of Therapy. Anticancer Res. 2015, 35, 5717–5735. [Google Scholar] [PubMed]
- Dei Tos, A.P.; Maestro, R.; Doglioni, C.; Gasparotto, D.; Boiocchi, M.; Laurino, L.; Fletcher, C.D. Ultraviolet-induced p53 mutations in atypical fibroxanthoma. Am. J. Pathol. 1994, 145, 11–17. [Google Scholar] [PubMed]
- Griewank, K.G.; Schilling, B.; Murali, R.; Bielefeld, N.; Schwamborn, M.; Sucker, A.; Zimmer, L.; Hillen, U.; Schaller, J.; Brenn, T.; et al. TERT promoter mutations are frequent in atypical fibroxanthomas and pleomorphic dermal sarcomas. Mod. Pathol. 2014, 27, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly recurrent TERT promoter mutations in human melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT promoter mutations in familial and sporadic melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Griewank, K.G.; Wiesner, T.; Murali, R.; Pischler, C.; Muller, H.; Koelsche, C.; Moller, I.; Franklin, C.; Cosgarea, I.; Sucker, A.; et al. Atypical fibroxanthoma and pleomorphic dermal sarcoma harbor frequent NOTCH1/2 and FAT1 mutations and similar DNA copy number alteration profiles. Mod. Pathol. 2018, 31, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Helbig, D.; Quaas, A.; Mauch, C.; Merkelbach-Bruse, S.; Buttner, R.; Emberger, M.; Wobser, M.; Russeler, V.; Putz, K.; Binot, E.; et al. Copy number variations in atypical fibroxanthomas and pleomorphic dermal sarcomas. Oncotarget 2017, 8, 109457–109467. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.; Quaas, A.; Noh, K.W.; Cartolano, M.; Abedpour, N.; Mauch, C.; Quantius, J.; Reinhardt, H.C.; Buettner, R.; Peifer, M.; et al. Integrative Analysis of Pleomorphic Dermal Sarcomas Reveals Fibroblastic Differentiation and Susceptibility to Immunotherapy. Clin. Cancer Res. 2020, 26, 5638–5645. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.L.; Randle, H.W.; Zalla, M.J.; Roenigk, R.K.; Brodland, D.G. A comparison of Mohs micrographic surgery and wide excision for the treatment of atypical fibroxanthoma. Dermatol. Surg. 1997, 23, 105–110. [Google Scholar] [CrossRef]
- Ang, G.C.; Roenigk, R.K.; Otley, C.C.; Kim Phillips, P.; Weaver, A.L. More than 2 decades of treating atypical fibroxanthoma at mayo clinic: What have we learned from 91 patients? Dermatol. Surg. 2009, 35, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Persa, O.D.; Loquai, C.; Wobser, M.; Baltaci, M.; Dengler, S.; Kreuter, A.; Volz, A.; Laimer, M.; Emberger, M.; Doerler, M.; et al. Extended surgical safety margins and ulceration are associated with an improved prognosis in pleomorphic dermal sarcomas. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 1577–1580. [Google Scholar] [CrossRef] [PubMed]
- Lonie, S.; Yau, B.; Henderson, M.; Gyorki, D.; Angel, C.; Webb, A. Management of pleomorphic dermal sarcoma. ANZ J. Surg. 2020, 90, 2322–2324. [Google Scholar] [CrossRef]
- Iorizzo, L.J.; Brown, M.D. Atypical fibroxanthoma: A review of the literature. Dermatol. Surg. 2011, 37, 146–157. [Google Scholar] [CrossRef]
- Miller, K.; Goodlad, J.R.; Brenn, T. Pleomorphic dermal sarcoma: Adverse histologic features predict aggressive behavior and allow distinction from atypical fibroxanthoma. Am. J. Surg. Pathol. 2012, 36, 1317–1326. [Google Scholar] [CrossRef]
- Tardío, J.C.; Pinedo, F.; Aramburu, J.A.; Suárez-Massa, D.; Pampín, A.; Requena, L.; Santonja, C. Pleomorphic dermal sarcoma: A more aggressive neoplasm than previously estimated. J. Cutan. Pathol. 2016, 43, 101–112. [Google Scholar] [CrossRef]
- Frampton, G.M.; Fichtenholtz, A.; Otto, G.A.; Wang, K.; Downing, S.R.; He, J.; Schnall-Levin, M.; White, J.; Sanford, E.M.; An, P.; et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 2013, 31, 1023–1031. [Google Scholar] [CrossRef]
- Anderson, H.L.; Joseph, A.K. A pilot feasibility study of a rare skin tumor database. Dermatol. Surg. 2007, 33, 693–696. [Google Scholar] [CrossRef]
- Cheng, K.A.; Kurtis, B.; Babayeva, S.; Zhuge, J.; Tantchou, I.; Cai, D.; Lafaro, R.J.; Fallon, J.T.; Zhong, M. Heterogeneity of TERT promoter mutations status in squamous cell carcinomas of different anatomical sites. Ann. Diagn. Pathol. 2015, 19, 146–148. [Google Scholar] [CrossRef]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [Green Version]
- Asada, S.; Fujino, T.; Goyama, S.; Kitamura, T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell. Mol. Life Sci. 2019, 76, 2511–2523. [Google Scholar] [CrossRef]
- Gao, L.; Hu, Y.; Tian, Y.; Fan, Z.; Wang, K.; Li, H.; Zhou, Q.; Zeng, G.; Hu, X.; Yu, L.; et al. Lung cancer deficient in the tumor suppressor GATA4 is sensitive to TGFBR1 inhibition. Nat. Commun. 2019, 10, 1665. [Google Scholar] [CrossRef]
- Lentjes, M.H.; Niessen, H.E.; Akiyama, Y.; de Bruïne, A.P.; Melotte, V.; van Engeland, M. The emerging role of GATA transcription factors in development and disease. Expert Rev. Mol. Med. 2016, 18, e3. [Google Scholar] [CrossRef]
- Martínez-Sáez, O.; Chic, N.; Pascual, T.; Adamo, B.; Vidal, M.; González-Farré, B.; Sanfeliu, E.; Schettini, F.; Conte, B.; Brasó-Maristany, F.; et al. Frequency and spectrum of PIK3CA somatic mutations in breast cancer. Breast Cancer Res. 2020, 22, 45. [Google Scholar] [CrossRef]
- Engelman, J.A. Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nat. Rev. Cancer 2009, 9, 550–562. [Google Scholar] [CrossRef]
- Samuels, Y.; Diaz, L.A.; Schmidt-Kittler, O.; Cummins, J.M.; Delong, L.; Cheong, I.; Rago, C.; Huso, D.L.; Lengauer, C.; Kinzler, K.W.; et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell 2005, 7, 561–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- André, F.; Ciruelos, E.M.; Juric, D.; Loibl, S.; Campone, M.; Mayer, I.A.; Rubovszky, G.; Yamashita, T.; Kaufman, B.; Lu, Y.S.; et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: Final overall survival results from SOLAR-1. Ann. Oncol. 2021, 32, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Bader, A.G.; Vogt, P.K. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc. Natl. Acad. Sci. USA 2005, 102, 802–807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meric-Bernstam, F.; Akcakanat, A.; Chen, H.; Do, K.A.; Sangai, T.; Adkins, F.; Gonzalez-Angulo, A.M.; Rashid, A.; Crosby, K.; Dong, M.; et al. PIK3CA/PTEN mutations and Akt activation as markers of sensitivity to allosteric mTOR inhibitors. Clin. Cancer Res. 2012, 18, 1777–1789. [Google Scholar] [CrossRef] [Green Version]
- Huang, G.; Liu, T.T.; Weng, S.W.; You, H.L.; Wei, Y.C.; Chen, C.H.; Eng, H.L.; Huang, W.T. CUL4A overexpression as an independent adverse prognosticator in intrahepatic cholangiocarcinoma. BMC Cancer 2017, 17, 395. [Google Scholar] [CrossRef] [Green Version]
- Mahmud, I.; Liao, D. DAXX in cancer: Phenomena, processes, mechanisms and regulation. Nucleic Acids Res. 2019, 47, 7734–7752. [Google Scholar] [CrossRef] [Green Version]
- Kilpivaara, O.; Levine, R.L. JAK2 and MPL mutations in myeloproliferative neoplasms: Discovery and science. Leukemia 2008, 22, 1813–1817. [Google Scholar] [CrossRef] [Green Version]
- Busch, E.L.; Hornick, J.L.; Umeton, R.; Albayrak, A.; Lindeman, N.I.; MacConaill, L.E.; Garcia, E.P.; Ducar, M.; Rebbeck, T.R. Somatic mutations in. Oncotarget 2017, 8, 85680–85691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Ahmadie, H.A.; Iyer, G.; Lee, B.H.; Scott, S.N.; Mehra, R.; Bagrodia, A.; Jordan, E.J.; Gao, S.P.; Ramirez, R.; Cha, E.K.; et al. Frequent somatic CDH1 loss-of-function mutations in plasmacytoid variant bladder cancer. Nat. Genet. 2016, 48, 356–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanza, F.; Maffini, E.; Rondoni, M.; Massari, E.; Faini, A.C.; Malavasi, F. CD22 Expression in B-Cell Acute Lymphoblastic Leukemia: Biological Significance and Implications for Inotuzumab Therapy in Adults. Cancers 2020, 12, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, V.K.; Gartner, J.J.; Samuels, Y.; Goldstein, A.M. The genetics of melanoma: Recent advances. Annu. Rev. Genom. Hum. Genet. 2013, 14, 257–279. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, G.P.; Denissenko, M.F.; Olivier, M.; Tretyakova, N.; Hecht, S.S.; Hainaut, P. Tobacco smoke carcinogens, DNA damage and p53 mutations in smoking-associated cancers. Oncogene 2002, 21, 7435–7451. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Chattopadhyay, R.; Okamura, R.; Saunders, I.M.; Montesion, M.; Frampton, G.M.; Miller, V.A.; Daniels, G.A.; Kurzrock, R. Phenotypic and Genomic Determinants of Immunotherapy Response Associated with Squamousness. Cancer Immunol. Res. 2019, 7, 866–873. [Google Scholar] [CrossRef] [Green Version]
- Painter, C.A.; Jain, E.; Tomson, B.N.; Dunphy, M.; Stoddard, R.E.; Thomas, B.S.; Damon, A.L.; Shah, S.; Kim, D.; Gómez Tejeda Zañudo, J.; et al. The Angiosarcoma Project: Enabling genomic and clinical discoveries in a rare cancer through patient-partnered research. Nat. Med. 2020, 26, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Persa, O.D.; Mauch, C.; Noh, K.W.; Pappesch, R.; Wagener-Ryczek, S.; Buettner, R.; Quaas, A.; Helbig, D. First report on two cases of pleomorphic dermal sarcoma successfully treated with immune checkpoint inhibitors. Oncoimmunology 2019, 8, e1665977. [Google Scholar] [CrossRef] [PubMed]
- Raedler, L.A. Keytruda (Pembrolizumab): First PD-1 Inhibitor Approved for Previously Treated Unresectable or Metastatic Melanoma. Am. Health Drug Benefits 2015, 8, 96–100. [Google Scholar] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| AFX | PDS | Total | |
|---|---|---|---|
| (n = 10) | (n = 13) | (n = 23) | |
| Diameter (cm) | |||
| ≤2 | 3 | 2 | 5 |
| 2.1–3.9 | 0 | 0 | 0 |
| ≥4.0 | 1 | 0 | 1 |
| Unknown | 6 | 11 | 17 |
| Invasion depth (mm) | |||
| ≥1 | 3 | 5 | 8 |
| ≤2 | 3 | 0 | 3 |
| 2.1–3.9 | 1 | 5 | 6 |
| ≥4.0 | 3 | 3 | 6 |
| Ulceration | |||
| Present | 5 | 7 | 12 |
| Absent | 5 | 6 | 11 |
| AFX | PDS | Total | |
|---|---|---|---|
| (n = 10) | (n = 18) | (n = 28) | |
| TMB-Status | |||
| High (≥10 mt/mb) | 10 | 17 | 27 |
| Low (1–9 mt/mb) | 0 | 1 | 1 |
| TMB (mt/mb) | |||
| Mean | 54.59 (20.2–90.8) | 69.49 (2.52–157.2) | 64.17 (2.52–157.2) |
| MSI-Status | |||
| Stable | 10 | 18 | 28 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ak, M.; Kahraman, A.; Arnold, F.M.; Turko, P.; Levesque, M.P.; Zoche, M.; Ramelyte, E.; Dummer, R. Clinicopathological and Genomic Profiles of Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma Identify Overlapping Signatures with a High Mutational Burden. Genes 2021, 12, 974. https://doi.org/10.3390/genes12070974
Ak M, Kahraman A, Arnold FM, Turko P, Levesque MP, Zoche M, Ramelyte E, Dummer R. Clinicopathological and Genomic Profiles of Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma Identify Overlapping Signatures with a High Mutational Burden. Genes. 2021; 12(7):974. https://doi.org/10.3390/genes12070974
Chicago/Turabian StyleAk, Melike, Abdullah Kahraman, Fabian M. Arnold, Patrick Turko, Mitchell P. Levesque, Martin Zoche, Egle Ramelyte, and Reinhard Dummer. 2021. "Clinicopathological and Genomic Profiles of Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma Identify Overlapping Signatures with a High Mutational Burden" Genes 12, no. 7: 974. https://doi.org/10.3390/genes12070974
APA StyleAk, M., Kahraman, A., Arnold, F. M., Turko, P., Levesque, M. P., Zoche, M., Ramelyte, E., & Dummer, R. (2021). Clinicopathological and Genomic Profiles of Atypical Fibroxanthoma and Pleomorphic Dermal Sarcoma Identify Overlapping Signatures with a High Mutational Burden. Genes, 12(7), 974. https://doi.org/10.3390/genes12070974