
Multiple Acyl-CoA Dehydrogenase Deficiency with Variable Presentation Due to a Homozygous Mutation in a Bedouin Tribe

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
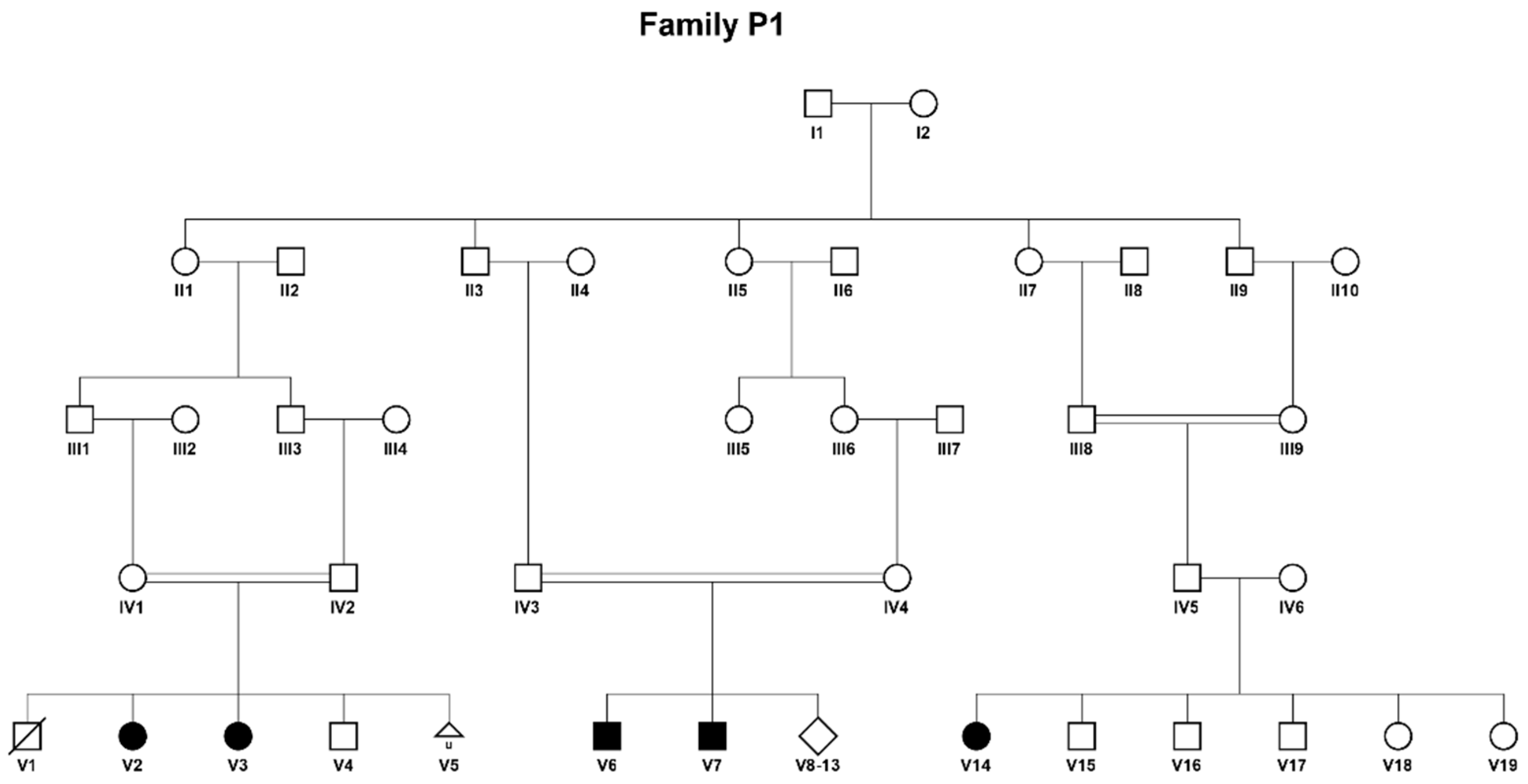
3.1. Clinical Studies
- Patient V1 (Figure 1) was the first son of consanguineous parents of Bedouin origin. He was born at term following an uneventful pregnancy with a birth weight of 3715 g and head circumference of 35 cm. The patient was discharged home after 2 days and died at home after two hours. Unfortunately, no investigations were performed.
- Patient V2 is a female, the second child in the same family. The pregnancy was uneventful, and she was born at term with a birth weight of 3570 g and head circumference of 34 cm. Apart from inspiratory stridor that resolved spontaneously, physical examination was considered normal. Due to the family history a metabolic work-up was performed immediately after birth (which was before the expanded newborn screening era in Israel) which resulted in the following findings: palmitate oxidation in lymphocytes was reduced to 40% in comparison to control, and acylcarnitine profile revealed increased levels of C4-C16 including C14 and C14:1, with the highest increase in C6-C10, accompanied by increased levels of glutarylcarnitine and isovalerylcarnitine (Table 1). These led to the suspected diagnosis of MADD. A urinary organic acids test at the time supported this clinical diagnosis. The patient was started on L-carnitine and riboflavin with normal development and no episodes of decompensation. The patient is now 16.5 years old and has been treated from birth with L-carnitine 50–100 mg/kg and riboflavin 100 mg.
- Patient V3 is a female, the third child in the same family. She was born at term after an uneventful pregnancy via cesarean section with a birth weight of 3610 g and head circumference of 36.5 cm. Newborn screening was positive twice and raised the suspicion of a MADD diagnosis, supported by a confirmatory metabolic work-up, including an acylcarnitine profile with increased C4-C10, C6 carnitines and upper range C5, C12-C18 and dicarboxylic aciduria with increased hexanoylglycine, glutaric, ethylmalonic and 2-OH-glutaric in urinary organic acids (Table 1). Due to suspected MADD, she was started on L-carnitine and riboflavin with normal development and no episodes of decompensation. The patient is now 10.5 years old and has been treated from birth with L-carnitine 50–100 mg/kg and riboflavin 100 mg.
- Patient V6 is a male, the first son in his nuclear family. He was born after an uneventful pregnancy to consanguineous parents of Bedouin origin with a birth weight of 4000 g and head circumference of 35 cm. At 9 months he was admitted due to encephalopathy after 4 days of diarrhea and refusal to feed and on physical examination had hepatomegaly, a micropenis and undescended testes. His viral panel, hepatitis A, B, and C serology, carcinoembryonic antigen (CEA) and α-fetoprotein were normal. Urine was negative for reducing substances. He was treated by intravenous D10 and electrolytes. On the fifth day, the patient deteriorated and developed hypoglycemia and hyperammonemia with elevated liver enzymes and normal serum bilirubin, prolonged coagulation tests and hyperlipidemia. A brain CT revealed mild frontal atrophy and ventriculomegaly. A liver biopsy demonstrated a Reye-like disease. Urinary organic acids showed increased glutaric- and β-OH-butyric acids. Total and free carnitine levels were low (Table 1). The patient improved with fresh frozen plasma, cryoprecipitate and total parenteral nutrition, and was started on L-carnitine with a diagnosis of fatty acid oxidation defect. At 16 years he developed recurrent perianal abscesses necessitating repeated drainage. The patient was treated from the age of 9 months until he was 18 years old with L-carnitine 50–100 mg/kg, though not consistently. He is currently 31 years old and is untreated due to lack of compliance.
- Patient V7 is a male, a sibling of patient V6 and the eighth child in the family. He was born after an uneventful pregnancy via cesarean section with a birth weight of 4280 g and head circumference of 37 cm. At 4 months he was admitted due to encephalopathy after 5 days of diarrhea and refusal to feed and on physical examination had hepatosplenomegaly accompanied by hypotonia. Laboratory work-up revealed hypoglycemia, elevated liver enzymes, elevated serum lactate, hypertriglyceridemia and thrombocytopenia. A viral panel and hepatitis A, B, and C serology were normal. Acylcarnitine profile showed a mild increase of C4–C10 and a marked increase of C12–C18, and urinary organic acids demonstrated massive excretion of dicarboxylic acids, increased lactic-, glutaric-, ethylmalonic-, p-OH-phenyl-lactic acids (Table 1). Total and free carnitine levels were low. He was treated by D10 and electrolyte infusion and was started on carnitine under a diagnosis of fatty acid oxidation defect. The patient has had recurrent admissions due to metabolic decompensations. He is currently 17 years old and has been treated from the age of 4 months with L-carnitine 100 mg/kg and later, due to low compliance, with 50 mg/kg.
- Patient V14 is a female, born at term after an uneventful pregnancy to consanguineous parents of Bedouin origin. She presented for the first time at the age of 19 years with muscle weakness, fatigue and abdominal pain for two months accompanied by weight loss. She was admitted due to inability to walk, rhabdomyolysis and elevated liver enzymes which resolved after D10 and electrolyte infusion, and due to suspected MADD was also started on high dose of L-carnitine and riboflavin. Metabolic work-up revealed increased C4–C16:1 in acylcarnitine profile and dicarboxylic aciduria with increased secretion of hexanoylglycine, glutaric-, ethylmalonic-, glutaric-, adipic-, isovaleric-, suberyl, 2-methyl butyry-acids on urinary organic acids profile (Table 1), which confirmed the diagnosis of MADD. The patient is 20 years old and has been treated with L-carnitine 50 mg/kg and riboflavin 100 mg for the last 14 months since her diagnosis and is currently stable with no admissions under this treatment.
3.2. Genetic Studies
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lindner, M.; Hoffmann, G.F.; Matern, D. Newborn screening for disorders of fatty-acid oxidation: Experience and recommendations from an expert meeting. J. Inherit. Metab. Dis. 2010, 33, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Xi, J.; Wen, B.; Lin, J.; Zhu, W.; Luo, S.; Zhao, C.; Li, D.; Lin, P.; Lu, J.; Yan, C. Clinical features and ETFDH mutation spectrum in a cohort of 90 chinese patients with late-onset multiple acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2014, 37, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Froissart, R.; Olsen, R.K.J.; Acquaviva, C.; Vianey-Saban, C. Electron transfer flavoprotein deficiency: Functional and molecular aspects. Mol. Genet. Metab. 2006, 88, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Frerman, F.E.; Goodman, S.I. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein:ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc. Natl. Acad. Sci. USA 1985, 82, 4517–4520. [Google Scholar] [CrossRef] [Green Version]
- Swigoňová, Z.; Mohsen, A.W.; Vockley, J. Acyl-coA dehydrogenases: Dynamic history of protein family evolution. J. Mol. Evol. 2009, 69, 176–193. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.K.J.; Andresen, B.S.; Christensen, E.; Bross, P.; Skovby, F.; Gregersen, N. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum. Mutat. 2003, 22, 12–23. [Google Scholar] [CrossRef]
- Law, L.K.; Tang, N.L.S.; Hui, J.; Fung, S.L.M.; Ruiter, J.; Wanders, R.J.A.; Fok, T.F.; Lam, C.W.K. Novel mutations in ETFDH gene in Chinese patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin. Chim. Acta 2009, 404, 95–99. [Google Scholar] [CrossRef]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3--new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- LF, F.; Chen, X.; Li, D.; Li, X.; Song, J.; Jin, Y.; Yang, Y. Reye syndrome and sudden death symptoms after oral administration of nimesulide due to upper respiratory tract infection in a boy. Zhongguo Dang Dai Er Ke Za Zhi 2018, 20, 944–949. [Google Scholar] [CrossRef]
- Fan, X.; Xie, B.; Zou, J.; Luo, J.; Qin, Z.; D’Gama, A.M.; Shi, J.; Yi, S.; Yang, Q.; Wang, J.; et al. Novel ETFDH mutations in four cases of riboflavin responsive multiple acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. Rep. 2018, 16, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Wen, B.; Li, D.; Shan, J.; Liu, S.; Li, W.; Zhao, Y.; Lin, P.; Zheng, J.; Li, D.; Gong, Y.; et al. Increased muscle coenzyme Q10 in riboflavin responsive MADD with ETFDH gene mutations due to secondary mitochondrial proliferation. Mol. Genet. Metab. 2013, 109, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Q.; Chen, X.J.; Murong, S.X.; Wang, N.; Wu, Z.Y. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A. J. Mol. Med. 2011, 89, 569–576. [Google Scholar] [CrossRef]
- Lan, M.Y.; Fu, M.H.; Liu, Y.F.; Huang, C.C.; Chang, Y.Y.; Liu, J.S.; Peng, C.H.; Chen, S.S. High frequency of ETFDH c.250G>A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin. Genet. 2010, 78, 565–569. [Google Scholar] [CrossRef]
- Yotsumoto, Y.; Hasegawa, Y.; Fukuda, S.; Kobayashi, H.; Endo, M.; Fukao, T.; Yamaguchi, S. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol. Genet. Metab. 2008, 94, 61–67. [Google Scholar] [CrossRef]
- Sim, N.-L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, a.D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Blanchette, M.; Kent, W.J.; Riemer, C.; Elnitski, L.; Smit, A.F.A.; Roskin, K.M.; Baertsch, R.; Rosenbloom, K.; Clawson, H.; Green, E.D. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004, 14, 708–715. [Google Scholar] [CrossRef] [Green Version]
- Ou, M.; Zhu, L.; Zhang, Y.; Zhang, Y.; Zhou, J.; Zhang, Y.; Chen, X.; Yang, L.; Li, T.; Su, X.; et al. A novel electron transfer flavoprotein dehydrogenase (ETFDH) gene mutation identified in a newborn with glutaric acidemia type II: A case report of a Chinese family. BMC Med. Genet. 2020, 21, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gempel, K.; Topaloglu, H.; Talim, B.; Schneiderat, P.; Schoser, B.G.H.; Hans, V.H.; Pálmafy, B.; Kale, G.; Tokatli, A.; Quinzii, C.; et al. The myopathic form of coenzyme Q10 deficiency is caused by mutations in the electron-transferring-flavoprotein dehydrogenase (ETFDH) gene. Brain 2007, 130, 2037–2044. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Pennisi, E.; Missaglia, S.; Tavian, D. Metabolic lipid muscle disorders: Biomarkers and treatment Corrado. Ther. Adv. Neurol. Disord. Rev. 2019, 12, 1–15. [Google Scholar] [CrossRef]
- Zhang, J.; Frerman, F.E.; Kim, J.J.P. Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. USA 2006, 103, 16212–16217. [Google Scholar] [CrossRef] [Green Version]
- Grünert, S.C. Clinical and genetical heterogeneity of late-onset multiple acyl-coenzyme A dehydrogenase deficiency. Orphanet J. Rare Dis. 2014, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Hazan, G.; Hershkovitz, E.; Staretz-Chacham, O. Incidence of inherited metabolic disorders in southern Israel: A comparison between consanguinity and non-consanguinity communities. Orphanet J. Rare Dis. 2020, 15, 1–7. [Google Scholar] [CrossRef]
- Missaglia, S.; Tavian, D.; Angelini, C. ETF dehydrogenase advances in molecular genetics and impact on treatment. Crit. Rev. Biochem. Mol. Biol. 2021, 56, 360–372. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.R.; Viau, K.; Botto, L.D.; Pasquali, M.; Longo, N. Clinical and biochemical outcomes of patients with medium-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2020, 129, 13–19. [Google Scholar] [CrossRef]
- Bleeker, J.C.; Kok, I.L.; Ferdinandusse, S.; van der Pol, W.L.; Cuppen, I.; Bosch, A.M.; Langeveld, M.; Derks, T.G.J.; Williams, M.; de Vries, M.; et al. Impact of newborn screening for very-long-chain acyl-CoA dehydrogenase deficiency on genetic, enzymatic, and clinical outcomes. J. Inherit. Metab. Dis. 2019, 42, 414–423. [Google Scholar] [CrossRef]
- Cornelius, N.; Frerman, F.E.; Corydon, T.J.; Palmfeldt, J.; Bross, P.; Gregersen, N.; Olsen, R.K.J. Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency. Hum. Mol. Genet. 2012, 21, 3435–3448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Er, T.K.; Chen, C.C.; Liu, Y.Y.; Chang, H.C.; Chien, Y.H.; Chang, J.G.; Hwang, J.K.; Jong, Y.J. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct. Biol. 2011, 11. [Google Scholar] [CrossRef] [Green Version]
- Olsen, R.K.J.; Olpin, S.E.; Andresen, B.S.; Miedzybrodzka, Z.H.; Pourfarzam, M.; Merinero, B.; Frerman, F.E.; Beresford, M.W.; Dean, J.C.S.; Cornelius, N.; et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain 2007, 130, 2045–2054. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Patient | Urinary Organic Acids | Acyl Carnitines | Free Carnitine (Norm: 25–45 mmol/L) | Total Carnitine (Norm: 30–50 mmol/L) |
|---|---|---|---|---|
| V2 | Increased hexanoylglycine, glutaric, ethylmalonic, 2-OH- glutaric | Increased C3/C5, C8, C10, C10:1, C12, C14, C14:1/C16 and glutarlcarnitine, isovalerylcarnitine | NA | NA |
| V3 | Dicarboxylic aciduria, mild ketonuria, increased hexanoylglycine, glutaric, ethylmalonic, 2-OH- glutaric | Increased C4, C6, C8, C10, C6 dicarboxylic; upper range C5, C12, C14, C16, C18 | 37.3 | 60.5 |
| V6 | NA | Mild increase C4–C18 | Low: 12 nmol/mL | NA |
| V7 | NA | Mild increase of C4–C10 and marked increase C12–C18 | Low: <10 nmol/mL | NA |
| V14 | Dicarboxylic aciduria, increased hexanoylglycine, glutaric, ethylmalonic, glutaric, adipic, isovaleryl, suberyl, 2-methyl butyryl | Increased C4, C6, C8, C10, C12, C14, C14:1, C14:2, C16, C16:1 | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Staretz-Chacham, O.; Amar, S.; Almashanu, S.; Pode-Shakked, B.; Saada, A.; Wormser, O.; Hershkovitz, E. Multiple Acyl-CoA Dehydrogenase Deficiency with Variable Presentation Due to a Homozygous Mutation in a Bedouin Tribe. Genes 2021, 12, 1140. https://doi.org/10.3390/genes12081140
Staretz-Chacham O, Amar S, Almashanu S, Pode-Shakked B, Saada A, Wormser O, Hershkovitz E. Multiple Acyl-CoA Dehydrogenase Deficiency with Variable Presentation Due to a Homozygous Mutation in a Bedouin Tribe. Genes. 2021; 12(8):1140. https://doi.org/10.3390/genes12081140
Chicago/Turabian StyleStaretz-Chacham, Orna, Shirly Amar, Shlomo Almashanu, Ben Pode-Shakked, Ann Saada, Ohad Wormser, and Eli Hershkovitz. 2021. "Multiple Acyl-CoA Dehydrogenase Deficiency with Variable Presentation Due to a Homozygous Mutation in a Bedouin Tribe" Genes 12, no. 8: 1140. https://doi.org/10.3390/genes12081140
APA StyleStaretz-Chacham, O., Amar, S., Almashanu, S., Pode-Shakked, B., Saada, A., Wormser, O., & Hershkovitz, E. (2021). Multiple Acyl-CoA Dehydrogenase Deficiency with Variable Presentation Due to a Homozygous Mutation in a Bedouin Tribe. Genes, 12(8), 1140. https://doi.org/10.3390/genes12081140