
DNA Polymerase θ: A Cancer Drug Target with Reverse Transcriptase Activity

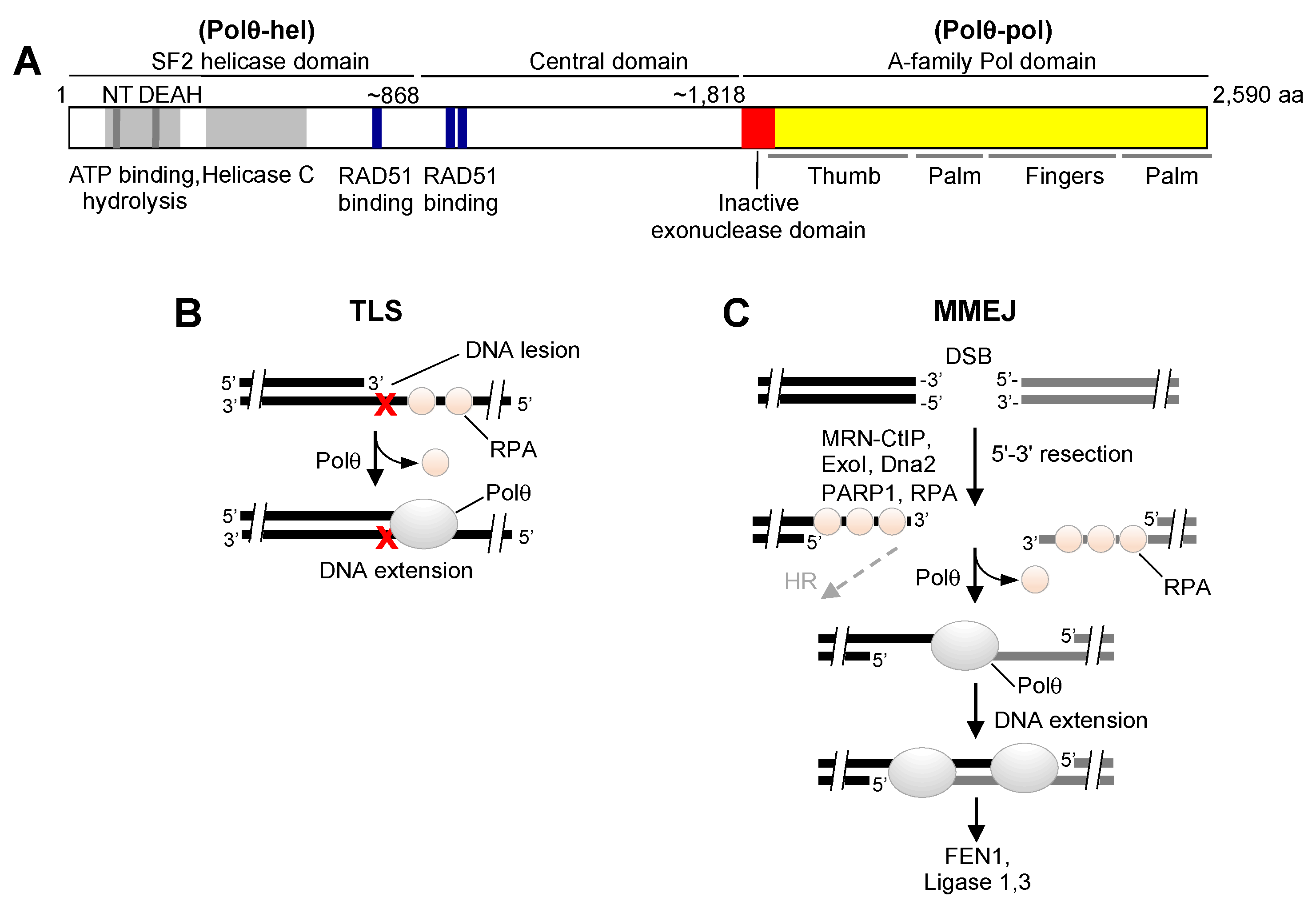{kind=link}
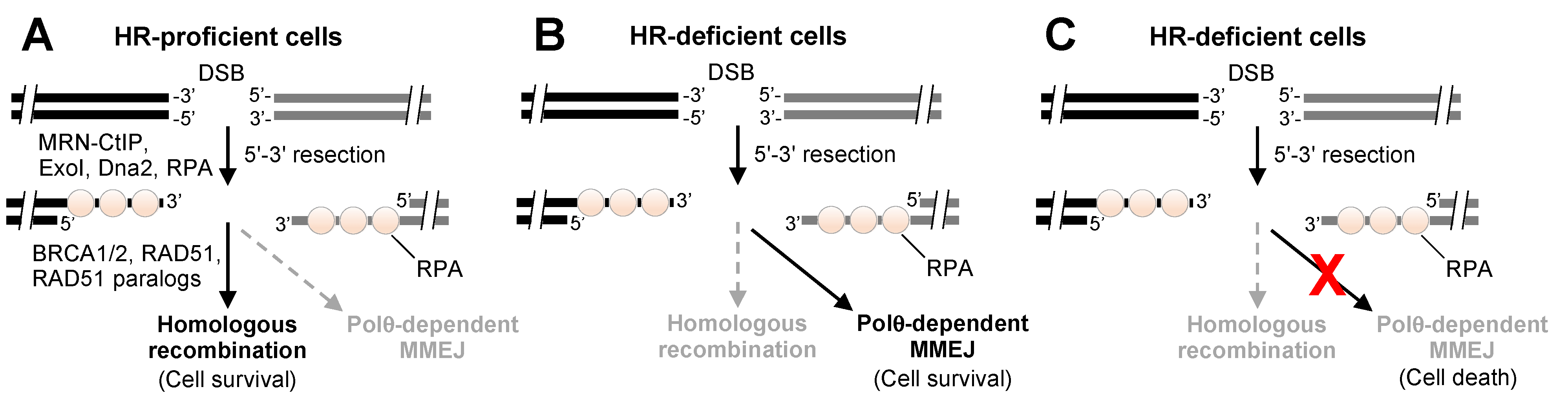{kind=link}
{kind=link}
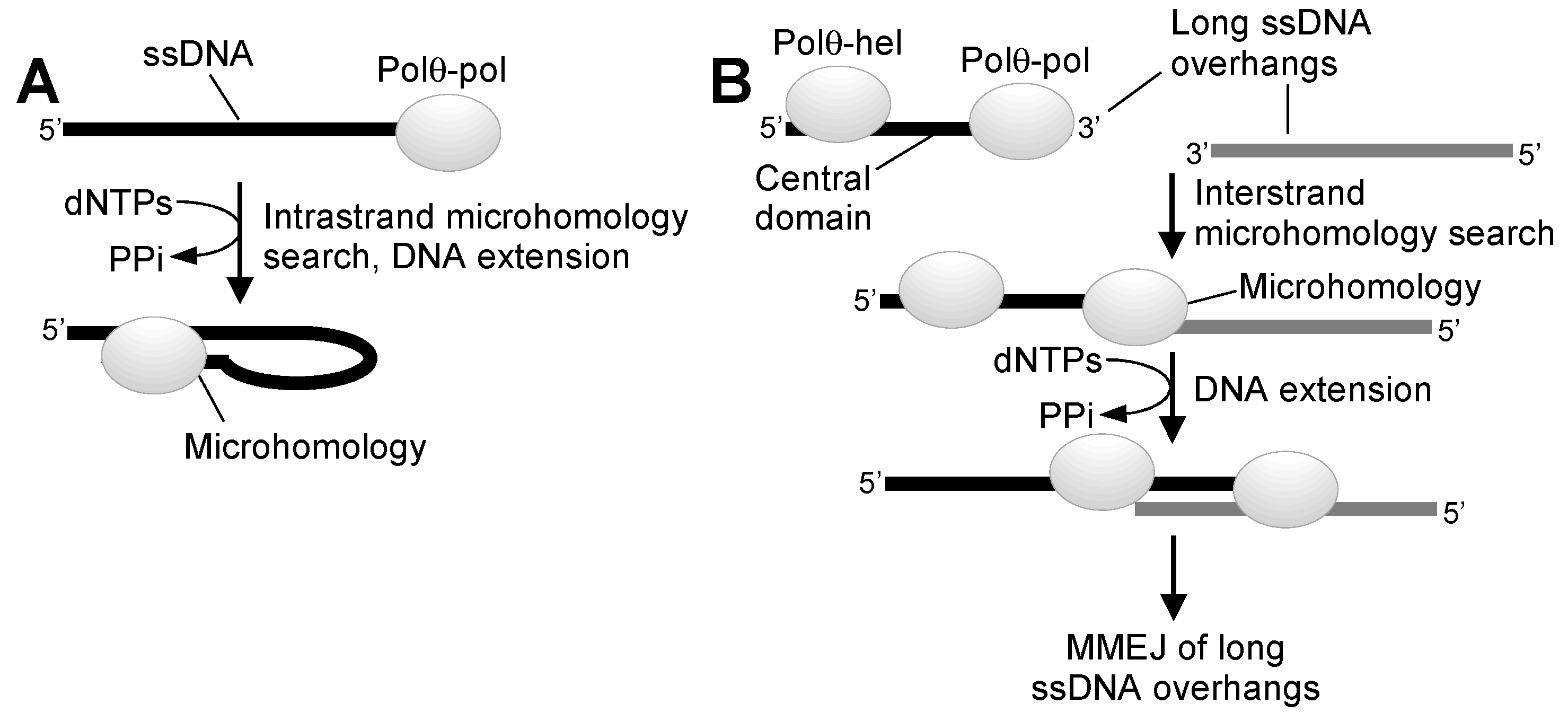{kind=link}
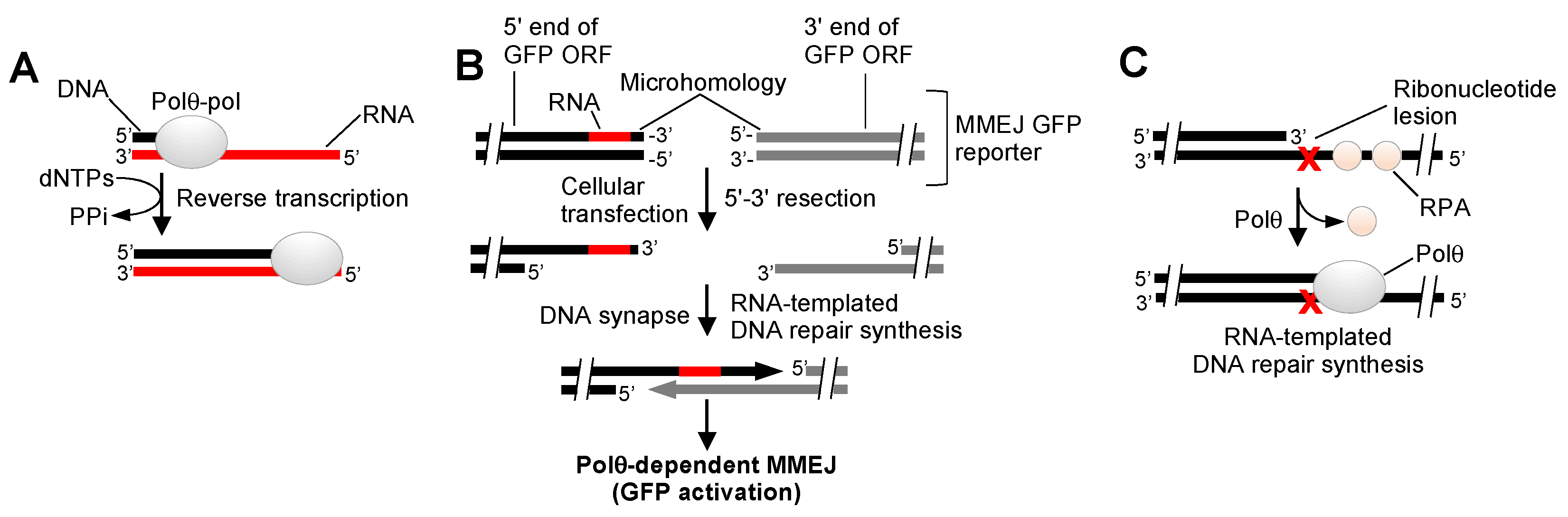{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Polθ DNA Repair Activities
3. Polθ Enzymatic Domains as Drug Targets
4. Polθ as a Reverse Transcriptase
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krais, J.J.; Johnson, N. BRCA1 Mutations in Cancer: Coordinating Deficiencies in Homologous Recombination with Tumorigenesis. Cancer Res. 2020, 80, 4601–4609. [Google Scholar] [CrossRef]
- Pan, Z.; Xie, X. BRCA mutations in the manifestation and treatment of ovarian cancer. Oncotarget 2017, 8, 97657–97670. [Google Scholar] [CrossRef] [Green Version]
- Couch, F.J.; Nathanson, K.L.; Offit, K. Two decades after BRCA: Setting paradigms in personalized cancer care and prevention. Science 2014, 343, 1466–1470. [Google Scholar] [CrossRef] [Green Version]
- Tutt, A.N.; Lord, C.J.; McCabe, N.; Farmer, H.; Turner, N.; Martin, N.M.; Jackson, S.P.; Smith, G.C.; Ashworth, A. Exploiting the DNA repair defect in BRCA mutant cells in the design of new therapeutic strategies for cancer. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 139–148. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [Green Version]
- Patel, P.S.; Algouneh, A.; Hakem, R. Exploiting synthetic lethality to target BRCA1/2-deficient tumors: Where we stand. Oncogene 2021, 40, 3001–3014. [Google Scholar] [CrossRef]
- Sonnenblick, A.; de Azambuja, E.; Azim, H.A.; Piccart, M. An update on PARP inhibitors--moving to the adjuvant setting. Nat. Rev. Clin. Oncol. 2015, 12, 27–41. [Google Scholar] [CrossRef]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef]
- Lord, C.J.; Ashworth, A. PARP inhibitors: Synthetic lethality in the clinic. Science 2017, 355, 1152–1158. [Google Scholar] [CrossRef]
- Noordermeer, S.M.; van Attikum, H. PARP Inhibitor Resistance: A Tug-of-War in BRCA-Mutated Cells. Trends Cell Biol. 2019, 29, 820–834. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, Z.Y.; Wu, N.; Chen, Y.C.; Cheng, Q.; Wang, J. PARP inhibitor resistance: The underlying mechanisms and clinical implications. Mol. Cancer 2020, 19, 107. [Google Scholar] [CrossRef] [PubMed]
- Mateos-Gomez, P.A.; Gong, F.; Nair, N.; Miller, K.M.; Lazzerini-Denchi, E.; Sfeir, A. Mammalian polymerase theta promotes alternative NHEJ and suppresses recombination. Nature 2015, 518, 254–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ceccaldi, R.; Liu, J.C.; Amunugama, R.; Hajdu, I.; Primack, B.; Petalcorin, M.I.R.; O’Connor, K.W.; Konstantinopoulos, P.A.; Elledge, S.J.; Boulton, S.J.; et al. Homologous-recombination-deficient tumours are dependent on Polθ-mediated repair. Nature 2015, 518, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Lemee, F.; Bergoglio, V.; Fernandez-Vidal, A.; Machado-Silva, A.; Pillaire, M.J.; Bieth, A.; Gentil, C.; Baker, L.; Martin, A.L.; Leduc, C.; et al. DNA polymerase theta up-regulation is associated with poor survival in breast cancer, perturbs DNA replication, and promotes genetic instability. Proc. Natl. Acad. Sci. USA 2010, 107, 13390–13395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, G.S.; Harris, A.L.; Prevo, R.; Helleday, T.; McKenna, W.G.; Buffa, F.M. Overexpression of POLQ confers a poor prognosis in early breast cancer patients. Oncotarget 2010, 1, 175–184. [Google Scholar] [CrossRef]
- Arana, M.E.; Seki, M.; Wood, R.D.; Rogozin, I.B.; Kunkel, T.A. Low-fidelity DNA synthesis by human DNA polymerase theta. Nucleic Acids Res. 2008, 36, 3847–3856. [Google Scholar] [CrossRef] [Green Version]
- Seki, M.; Masutani, C.; Yang, L.W.; Schuffert, A.; Iwai, S.; Bahar, I.; Wood, R.D. High-efficiency bypass of DNA damage by human DNA polymerase Q. EMBO J. 2004, 23, 4484–4494. [Google Scholar] [CrossRef] [Green Version]
- Begg, A. POLQ in breast cancer. Oncotarget 2010, 1, 161–162. [Google Scholar] [CrossRef]
- Chandramouly, G.; Liao, S.; Rusanov, T.; Borisonnik, N.; Calbert, M.L.; Kent, T.; Sullivan-Reed, K.; Vekariya, U.; Kashkina, E.; Skorski, T.; et al. Poltheta promotes the repair of 5′-DNA-protein crosslinks by microhomology-mediated end-joining. Cell Rep. 2021, 34, 108820. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Wyatt, D.W.; Takata, K.; Mu, Y.; Hensley, S.C.; Tomida, J.; Bylund, G.O.; Doublie, S.; Johansson, E.; Ramsden, D.A.; et al. Mechanism of suppression of chromosomal instability by DNA polymerase POLQ. PLoS Genet. 2014, 10, e1004654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, G.S.; Prevo, R.; Lee, Y.F.; Helleday, T.; Muschel, R.J.; Taylor, S.; Yoshimura, M.; Hickson, I.D.; Bernhard, E.J.; McKenna, W.G. A small interfering RNA screen of genes involved in DNA repair identifies tumor-specific radiosensitization by POLQ knockdown. Cancer Res. 2010, 70, 2984–2993. [Google Scholar] [CrossRef] [Green Version]
- Dai, C.H.; Chen, P.; Li, J.; Lan, T.; Chen, Y.C.; Qian, H.; Chen, K.; Li, M.Y. Co-inhibition of pol theta and HR genes efficiently synergize with cisplatin to suppress cisplatin-resistant lung cancer cells survival. Oncotarget 2016, 7, 65157–65170. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.A.; Cooper, C.D.O.; Aitkenhead, H.; Gileadi, O. Structure of the Helicase Domain of DNA Polymerase Theta Reveals a Possible Role in the Microhomology-Mediated End-Joining Pathway. Structure 2015, 23, 2319–2330. [Google Scholar] [CrossRef] [Green Version]
- Black, S.J.; Ozdemir, A.Y.; Kashkina, E.; Kent, T.; Rusanov, T.; Ristic, D.; Shin, Y.; Suma, A.; Hoang, T.; Chandramouly, G.; et al. Molecular basis of microhomology-mediated end-joining by purified full-length Poltheta. Nat. Commun. 2019, 10, 4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahn, K.E.; Averill, A.M.; Aller, P.; Wood, R.D.; Doublie, S. Human DNA polymerase theta grasps the primer terminus to mediate DNA repair. Nat. Struct. Mol. Biol. 2015, 22, 304–311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, M.; Marini, F.; Wood, R.D. POLQ (Pol theta), a DNA polymerase and DNA-dependent ATPase in human cells. Nucleic Acids Res. 2003, 31, 6117–6126. [Google Scholar] [CrossRef] [Green Version]
- Kent, T.; Rusanov, T.D.; Hoang, T.M.; Velema, W.A.; Krueger, A.T.; Copeland, W.C.; Kool, E.T.; Pomerantz, R.T. DNA polymerase theta specializes in incorporating synthetic expanded-size (xDNA) nucleotides. Nucleic Acids Res. 2016, 44, 9381–9392. [Google Scholar] [CrossRef] [Green Version]
- Kent, T.; Mateos-Gomez, P.A.; Sfeir, A.; Pomerantz, R.T. Polymerase theta is a robust terminal transferase that oscillates between three different mechanisms during end-joining. eLife 2016, 5, e13740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seki, M.; Wood, R.D. DNA polymerase theta (POLQ) can extend from mismatches and from bases opposite a (6-4) photoproduct. DNA Repair 2008, 7, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Hogg, M.; Seki, M.; Wood, R.D.; Doublie, S.; Wallace, S.S. Lesion bypass activity of DNA polymerase theta (POLQ) is an intrinsic property of the pol domain and depends on unique sequence inserts. J. Mol. Biol. 2011, 405, 642–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; Roy Choudhury, J.; Park, J.; Prakash, S.; Prakash, L. A Role for DNA Polymerase theta in Promoting Replication through Oxidative DNA Lesion, Thymine Glycol, in Human Cells. J. Biol. Chem. 2014, 289, 13177–13185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.H.; McArthur, M.J.; Park, J.; Basu, D.; Wakamiya, M.; Prakash, L.; Prakash, S. Error-Prone Replication through UV Lesions by DNA Polymerase theta Protects against Skin Cancers. Cell 2019, 176, 1295–1309. [Google Scholar] [CrossRef] [Green Version]
- Arana, M.E.; Takata, K.; Garcia-Diaz, M.; Wood, R.D.; Kunkel, T.A. A unique error signature for human DNA polymerase nu. DNA Repair 2007, 6, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Jain, R.; Aggarwal, A.K.; Rechkoblit, O. Eukaryotic DNA polymerases. Curr. Opin. Struct. Biol. 2018, 53, 77–87. [Google Scholar] [CrossRef]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef] [Green Version]
- Koole, W.; van Schendel, R.; Karambelas, A.E.; van Heteren, J.T.; Okihara, K.L.; Tijsterman, M. A Polymerase Theta-dependent repair pathway suppresses extensive genomic instability at endogenous G4 DNA sites. Nat. Commun. 2014, 5, 3216. [Google Scholar] [CrossRef] [Green Version]
- Wyatt, D.W.; Feng, W.; Conlin, M.P.; Yousefzadeh, M.J.; Roberts, S.A.; Mieczkowski, P.; Wood, R.D.; Gupta, G.P.; Ramsden, D.A. Essential Roles for Polymerase theta-Mediated End Joining in the Repair of Chromosome Breaks. Mol. Cell 2016, 63, 662–673. [Google Scholar] [CrossRef] [Green Version]
- Zahn, K.E.; Jensen, R.B.; Wood, R.D.; Doublie, S. Human DNA polymerase theta harbors DNA end-trimming activity critical for DNA repair. Mol. Cell 2021, 81, 1534–1547.e4. [Google Scholar] [CrossRef] [PubMed]
- Prasad, R.; Longley, M.J.; Sharief, F.S.; Hou, E.W.; Copeland, W.C.; Wilson, S.H. Human DNA polymerase theta possesses 5’-dRP lyase activity and functions in single-nucleotide base excision repair in vitro. Nucleic Acids Res. 2009, 37, 1868–1877. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Song, Y.; Li, S.; Kurian, S.; Xiang, R.; Chiba, T.; Wu, X. DNA polymerase theta (POLQ) is important for repair of DNA double-strand breaks caused by fork collapse. J. Biol. Chem. 2019, 294, 3909–3919. [Google Scholar] [CrossRef]
- Shima, N.; Munroe, R.J.; Schimenti, J.C. The mouse genomic instability mutation chaos1 is an allele of Polq that exhibits genetic interaction with Atm. Mol. Cell Biol. 2004, 24, 10381–10389. [Google Scholar] [CrossRef] [Green Version]
- Carvajal-Garcia, J.; Crown, K.N.; Ramsden, D.A.; Sekelsky, J. DNA polymerase theta suppresses mitotic crossing over. PLoS Genet. 2021, 17, e1009267. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Khoo, K.J.; Zhang, Y.; Maizels, N. POLQ suppresses interhomolog recombination and loss of heterozygosity at targeted DNA breaks. Proc. Natl. Acad. Sci. USA 2020, 117, 22900–22909. [Google Scholar] [CrossRef]
- Moynahan, M.E.; Jasin, M. Mitotic homologous recombination maintains genomic stability and suppresses tumorigenesis. Nat. Rev. Mol. Cell Biol. 2010, 11, 196–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [Green Version]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Jasin, M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef]
- Cho, N.W.; Greenberg, R.A. DNA repair: Familiar ends with alternative endings. Nature 2015, 518, 174–176. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic Screens Reveal FEN1 and APEX2 as BRCA2 Synthetic Lethal Targets. Mol. Cell 2019, 73, 885–899.e886. [Google Scholar] [CrossRef] [Green Version]
- Mateos-Gomez, P.A.; Kent, T.; Deng, S.K.; McDevitt, S.; Kashkina, E.; Hoang, T.M.; Pomerantz, R.T.; Sfeir, A. The helicase domain of Poltheta counteracts RPA to promote alt-NHEJ. Nat. Struct. Mol. Biol. 2017, 24, 1116–1123. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef] [Green Version]
- Zatreanu, D.; Robinson, H.M.R.; Alkhatib, O.; Boursier, M.; Finch, H.; Geo, L.; Grande, D.; Grinkevich, V.; Heald, R.A.; Langdon, S.; et al. Poltheta inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat. Commun. 2021, 12, 3636. [Google Scholar] [CrossRef]
- Ozdemir, A.Y.; Rusanov, T.; Kent, T.; Siddique, L.A.; Pomerantz, R.T. Polymerase theta-helicase efficiently unwinds DNA and RNA-DNA hybrids. J. Biol. Chem. 2018, 293, 5259–5269. [Google Scholar] [CrossRef] [Green Version]
- Deng, S.K.; Gibb, B.; de Almeida, M.J.; Greene, E.C.; Symington, L.S. RPA antagonizes microhomology-mediated repair of DNA double-strand breaks. Nat. Struct. Mol. Biol. 2014, 21, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Gelot, C.; Pantelidou, C.; Li, A.; Yucel, H.; Davis, R.E.; Farkkila, A.; Kochupurakkal, B.; Syed, A.; Shapiro, G.I.; et al. A first-in-class Polymerase Theta Inhibitor selectively targets Homologous-Recombination-Deficient Tumors. Nat. Cancer 2021, 2, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Schrempf, A.; Slyskova, J.; Loizou, J.I. Targeting the DNA Repair Enzyme Polymerase θ in Cancer Therapy. Trends Cancer 2020, 7, 98–111. [Google Scholar] [CrossRef]
- Zeng, S.; Huang, W.; Zheng, X.; Liyan, C.; Zhang, Z.; Wang, J.; Shen, Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. Eur. J. Med. Chem. 2021, 210, 112981. [Google Scholar] [CrossRef] [PubMed]
- Paiva, S.L.; Crews, C.M. Targeted protein degradation: Elements of PROTAC design. Curr. Opin. Chem. Biol. 2019, 50, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Gao, H.; Yang, Y.; He, M.; Wu, Y.; Song, Y.; Tong, Y.; Rao, Y. PROTACs: Great opportunities for academia and industry. Signal Transduct. Target. Ther. 2019, 4, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, C.; Shen, X.; Niu, S.; Ma, C. Innate Reverse Transcriptase Activity of DNA Polymerase for Isothermal RNA Direct Detection. J. Am. Chem. Soc. 2015, 137, 13804–13806. [Google Scholar] [CrossRef] [PubMed]
- Menendez-Arias, L.; Sebastian-Martin, A.; Alvarez, M. Viral reverse transcriptases. Virus Res. 2017, 234, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Chandramouly, G.; Zhao, J.; McDevitt, S.; Rusanov, T.; Hoang, T.; Borisonnik, N.; Treddinick, T.; Lopezcolorado, F.W.; Kent, T.; Siddique, L.A.; et al. Poltheta reverse transcribes RNA and promotes RNA-templated DNA repair. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Korolev, S.; Waksman, G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: Structural basis for nucleotide incorporation. EMBO J. 1998, 17, 7514–7525. [Google Scholar] [CrossRef] [Green Version]
- Doublie, S.; Sawaya, M.R.; Ellenberger, T. An open and closed case for all polymerases. Structure 1999, 7, R31–R35. [Google Scholar] [CrossRef] [Green Version]
- Miller III, B.R.; Beese, L.S.; Parish, C.A.; Wu, E.Y. The Closing Mechanism of DNA Polymerase I at Atomic Resolution. Structure 2015, 23, 1609–1620. [Google Scholar] [CrossRef] [Green Version]
- Kiefer, J.R.; Mao, C.; Braman, J.C.; Beese, L.S. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature 1998, 391, 304–307. [Google Scholar] [CrossRef]
- Chim, N.; Meza, R.A.; Trinh, A.M.; Yang, K.; Chaput, J.C. Following replicative DNA synthesis by time-resolved X-ray crystallography. Nat. Commun. 2021, 12, 2641. [Google Scholar] [CrossRef]
- Nowak, E.; Potrzebowski, W.; Konarev, P.V.; Rausch, J.W.; Bona, M.K.; Svergun, D.I.; Bujnicki, J.M.; Le Grice, S.F.; Nowotny, M. Structural analysis of monomeric retroviral reverse transcriptase in complex with an RNA/DNA hybrid. Nucleic Acids Res. 2013, 41, 3874–3887. [Google Scholar] [CrossRef] [Green Version]
- Sarafianos, S.G.; Das, K.; Tantillo, C.; Clark, A.D., Jr.; Ding, J.; Whitcomb, J.M.; Boyer, P.L.; Hughes, S.H.; Arnold, E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001, 20, 1449–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelini, F.; Pitchiaya, S.; Vitelli, V.; Sharma, S.; Gioia, U.; Pessina, F.; Cabrini, M.; Wang, Y.; Capozzo, I.; Iannelli, F.; et al. Damage-induced lncRNAs control the DNA damage response through interaction with DDRNAs at individual double-strand breaks. Nat. Cell Biol. 2017, 19, 1400–1411. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Ba, Z.; Gao, M.; Wu, Y.; Ma, Y.; Amiard, S.; White, C.I.; Rendtlew Danielsen, J.M.; Yang, Y.G.; Qi, Y. A role for small RNAs in DNA double-strand break repair. Cell 2012, 149, 101–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, S.; Anand, R.; Zhang, X.; Francia, S.; Michelini, F.; Galbiati, A.; Williams, H.; Ronato, D.A.; Masson, J.Y.; Rothenberg, E.; et al. MRE11-RAD50-NBS1 Complex Is Sufficient to Promote Transcription by RNA Polymerase II at Double-Strand Breaks by Melting DNA Ends. Cell Rep. 2021, 34, 108565. [Google Scholar] [CrossRef]
- Mazina, O.M.; Keskin, H.; Hanamshet, K.; Storici, F.; Mazin, A.V. Rad52 Inverse Strand Exchange Drives RNA-Templated DNA Double-Strand Break Repair. Mol. Cell 2017, 67, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meers, C.; Keskin, H.; Banyai, G.; Mazina, O.; Yang, T.; Gombolay, A.L.; Mukherjee, K.; Kaparos, E.I.; Newnam, G.; Mazin, A.; et al. Genetic Characterization of Three Distinct Mechanisms Supporting RNA-Driven DNA Repair and Modification Reveals Major Role of DNA Polymerase zeta. Mol. Cell 2020, 79, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Lazzaro, F.; Novarina, D.; Amara, F.; Watt, D.L.; Stone, J.E.; Costanzo, V.; Burgers, P.M.; Kunkel, T.A.; Plevani, P.; Muzi-Falconi, M. RNase H and postreplication repair protect cells from ribonucleotides incorporated in DNA. Mol. Cell 2012, 45, 99–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.S.; Kunkel, T.A. Ribonucleotides in DNA: Origins, repair and consequences. DNA Repair 2014, 19, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, J.S.; Lujan, S.A.; Kunkel, T.A. Processing ribonucleotides incorporated during eukaryotic DNA replication. Nat. Rev. Mol. Cell Biol. 2016, 17, 350–363. [Google Scholar] [CrossRef] [Green Version]
- Cihlar, T.; Ray, A.S. Nucleoside and nucleotide HIV reverse transcriptase inhibitors: 25 years after zidovudine. Antivir. Res. 2010, 85, 39–58. [Google Scholar] [CrossRef]
- Mehellou, Y.; Rattan, H.S.; Balzarini, J. The ProTide Prodrug Technology: From the Concept to the Clinic. J. Med. Chem. 2018, 61, 2211–2226. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Durantel, D.; Zoulim, F.; Dumontet, C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 2013, 12, 447–464. [Google Scholar] [CrossRef] [PubMed]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef] [Green Version]
- Namasivayam, V.; Vanangamudi, M.; Kramer, V.G.; Kurup, S.; Zhan, P.; Liu, X.; Kongsted, J.; Byrareddy, S.N. The Journey of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) from Lab to Clinic. J. Med. Chem. 2019, 62, 4851–4883. [Google Scholar] [CrossRef] [PubMed]
- Kokic, G.; Hillen, H.S.; Tegunov, D.; Dienemann, C.; Seitz, F.; Schmitzova, J.; Farnung, L.; Siewert, A.; Hobartner, C.; Cramer, P. Mechanism of SARS-CoV-2 polymerase stalling by remdesivir. Nat. Commun. 2021, 12, 279. [Google Scholar] [CrossRef]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020, 382, 2327–2336. [Google Scholar] [CrossRef]
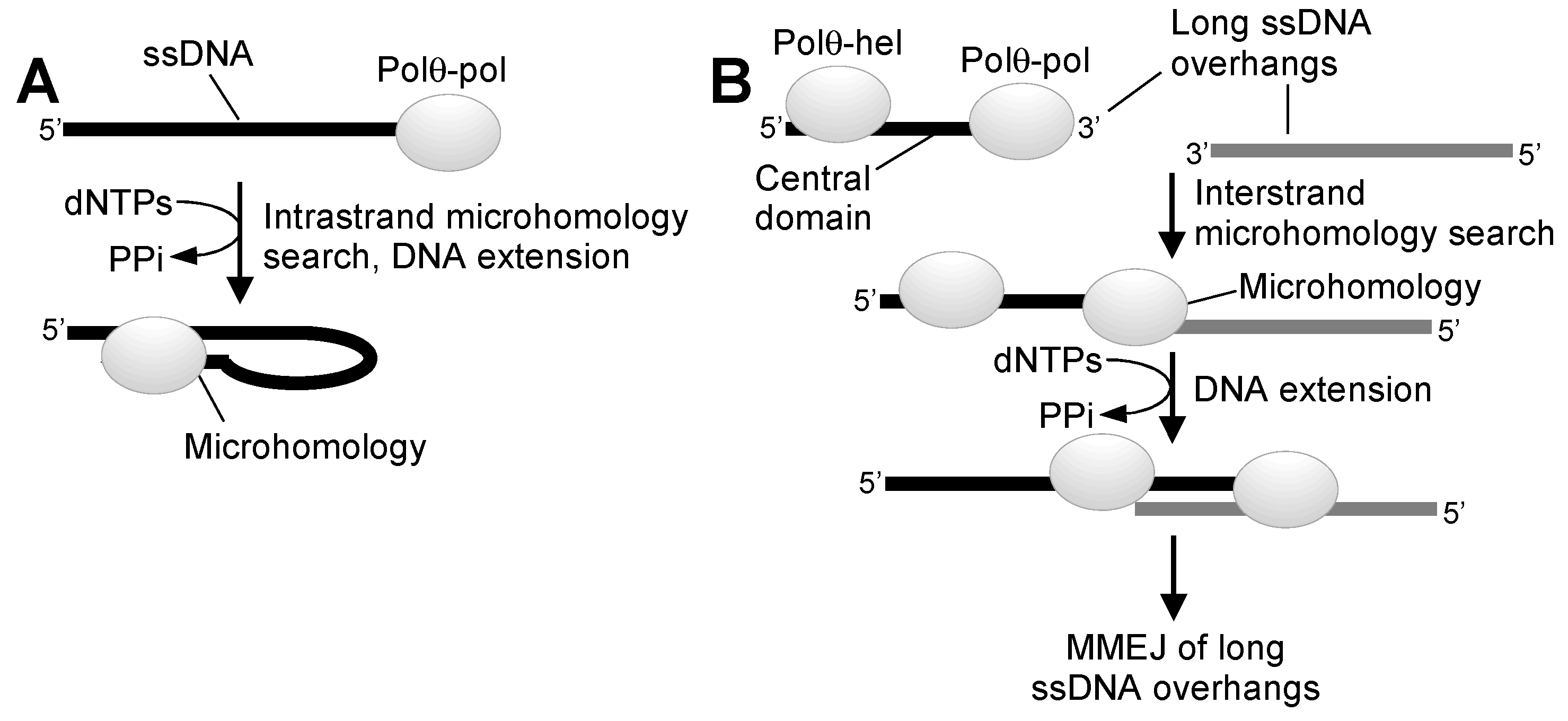

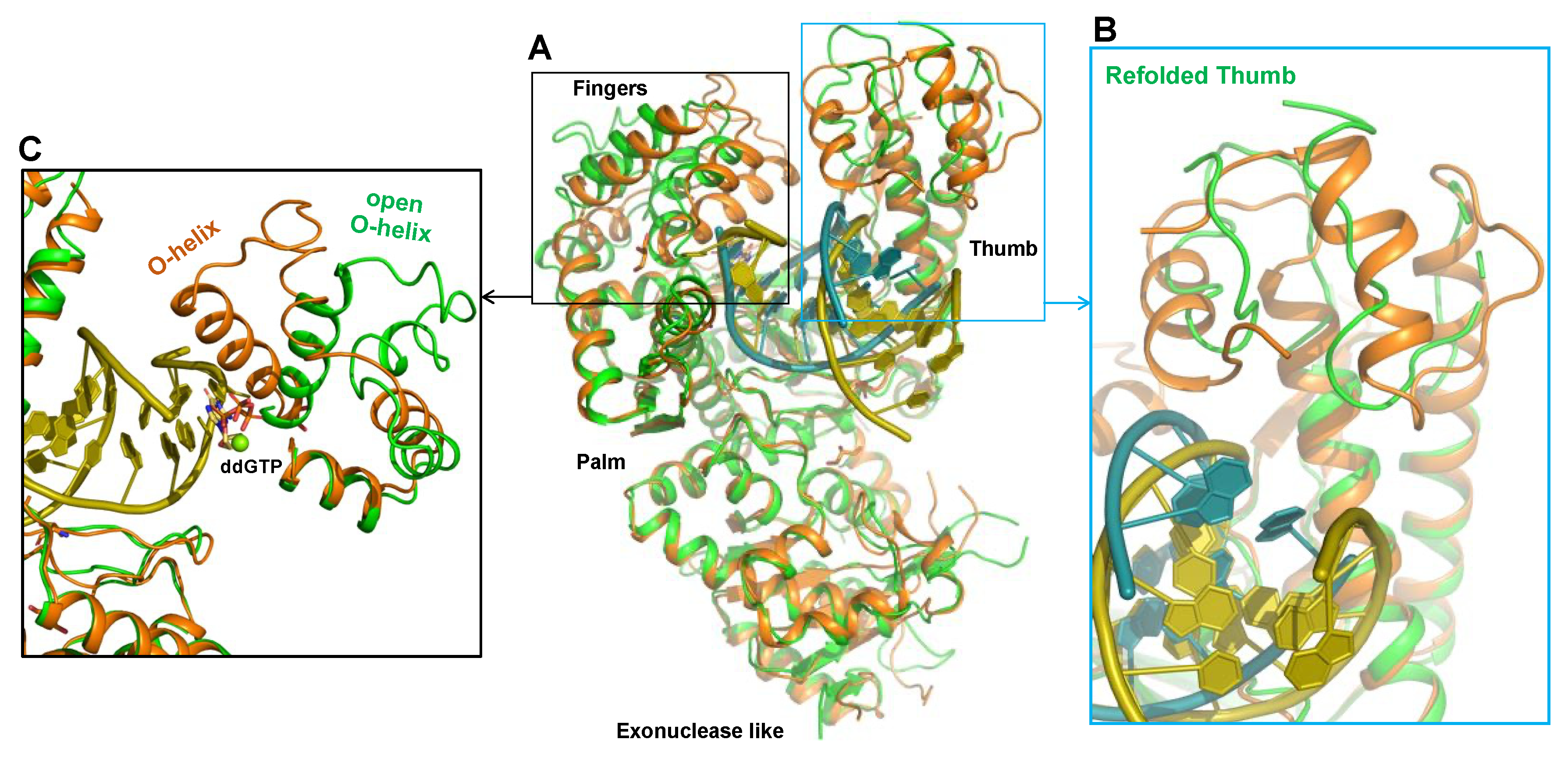
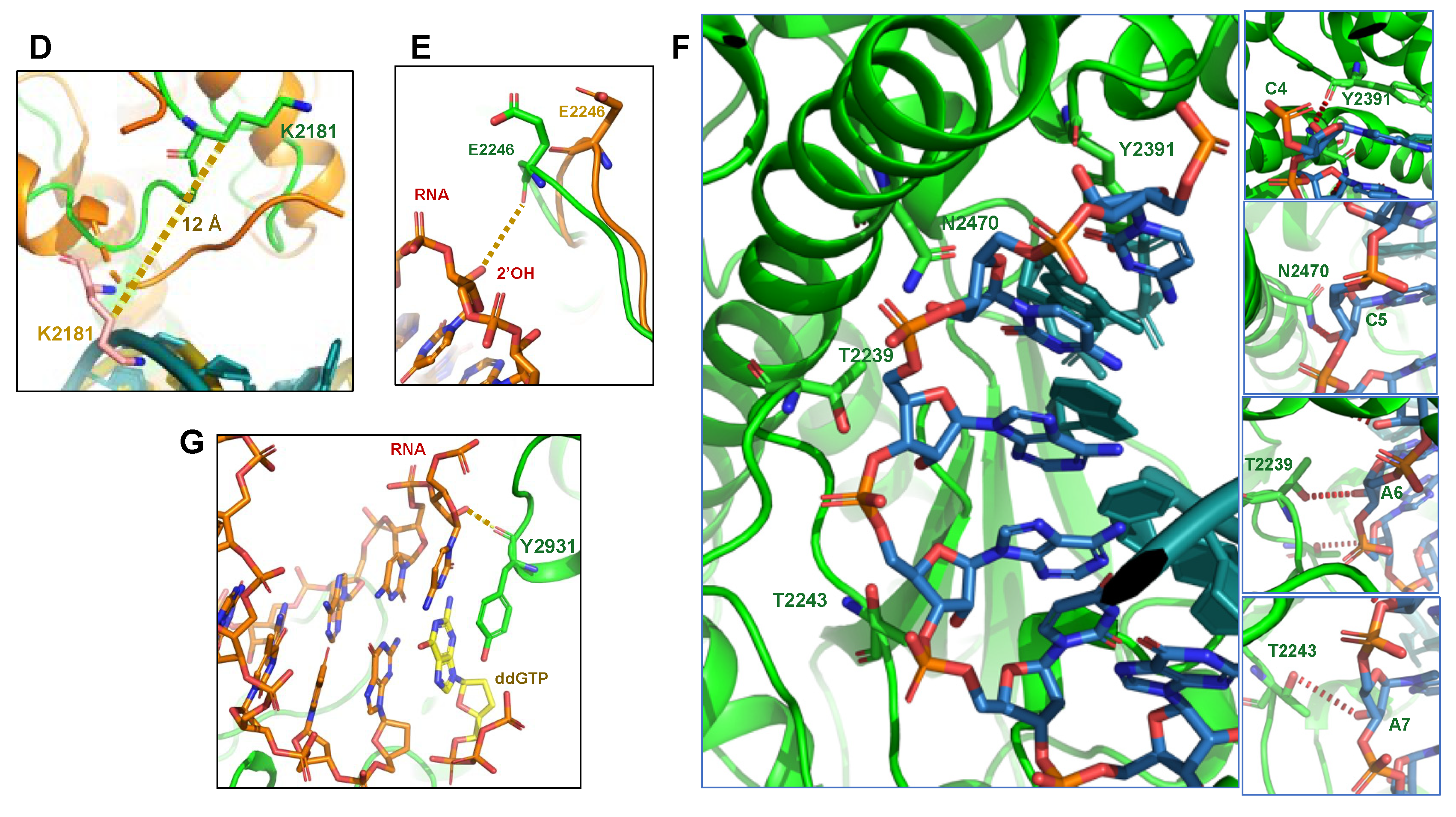
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.S.; Pomerantz, R.T. DNA Polymerase θ: A Cancer Drug Target with Reverse Transcriptase Activity. Genes 2021, 12, 1146. https://doi.org/10.3390/genes12081146
Chen XS, Pomerantz RT. DNA Polymerase θ: A Cancer Drug Target with Reverse Transcriptase Activity. Genes. 2021; 12(8):1146. https://doi.org/10.3390/genes12081146
Chicago/Turabian StyleChen, Xiaojiang S., and Richard T. Pomerantz. 2021. "DNA Polymerase θ: A Cancer Drug Target with Reverse Transcriptase Activity" Genes 12, no. 8: 1146. https://doi.org/10.3390/genes12081146
APA StyleChen, X. S., & Pomerantz, R. T. (2021). DNA Polymerase θ: A Cancer Drug Target with Reverse Transcriptase Activity. Genes, 12(8), 1146. https://doi.org/10.3390/genes12081146