
Systematic Identification of circRNAs in Alzheimer’s Disease

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
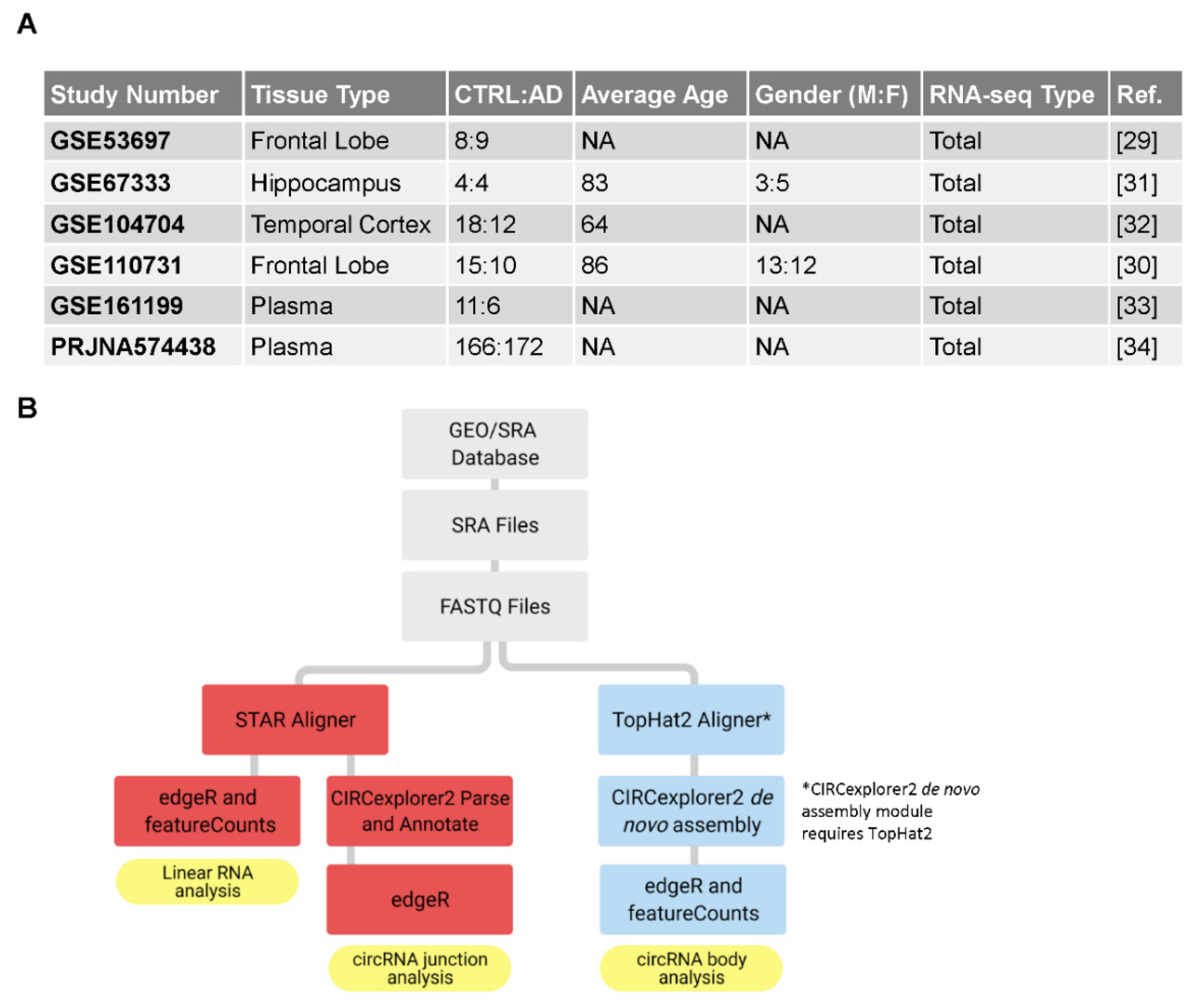
2.1. RNA Sequencing Datasets from Brain Tissue and Plasma
2.2. Aligning FASTQ Files to the Human Genome
2.3. Using CIRCexplorer2 to Analyze Junction Reads
2.4. Constructing circRNA Body Approximations with De Novo Assembly
2.5. Gene Expression Analysis in R
2.6. Relating circRNAs to AD Pathology
3. Results
3.1. Analysis of AD-Associated RNA-Seq Datasets to Identify circRNAs
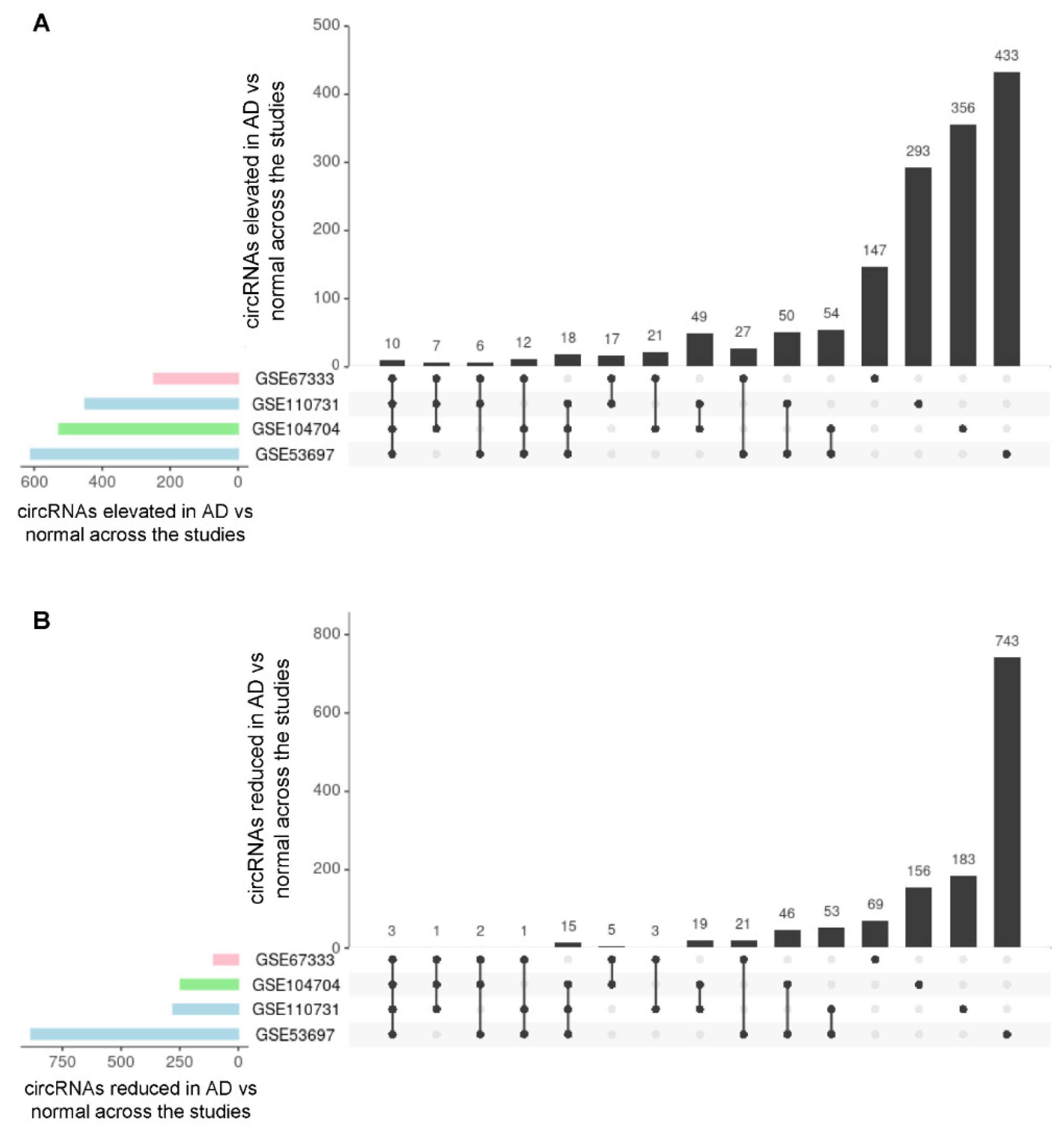
3.2. Analysis of circRNAs Differentially Abundant in AD Brain Compared with Control Brain
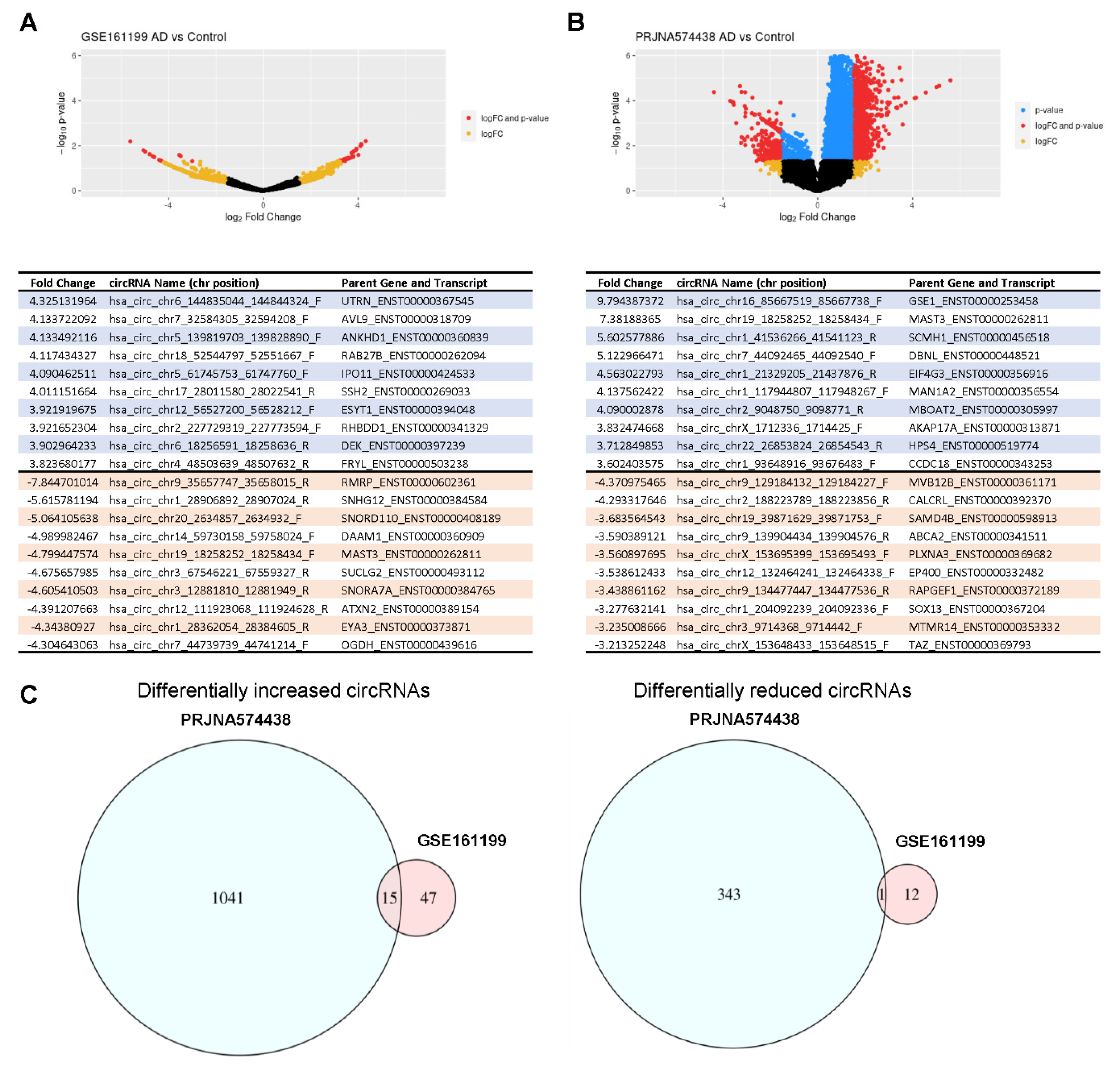
3.3. Analysis of circRNAs Differentially Abundant in AD Plasma Compared with Control Plasma
3.4. Predictions of Full circRNA Body Sequences
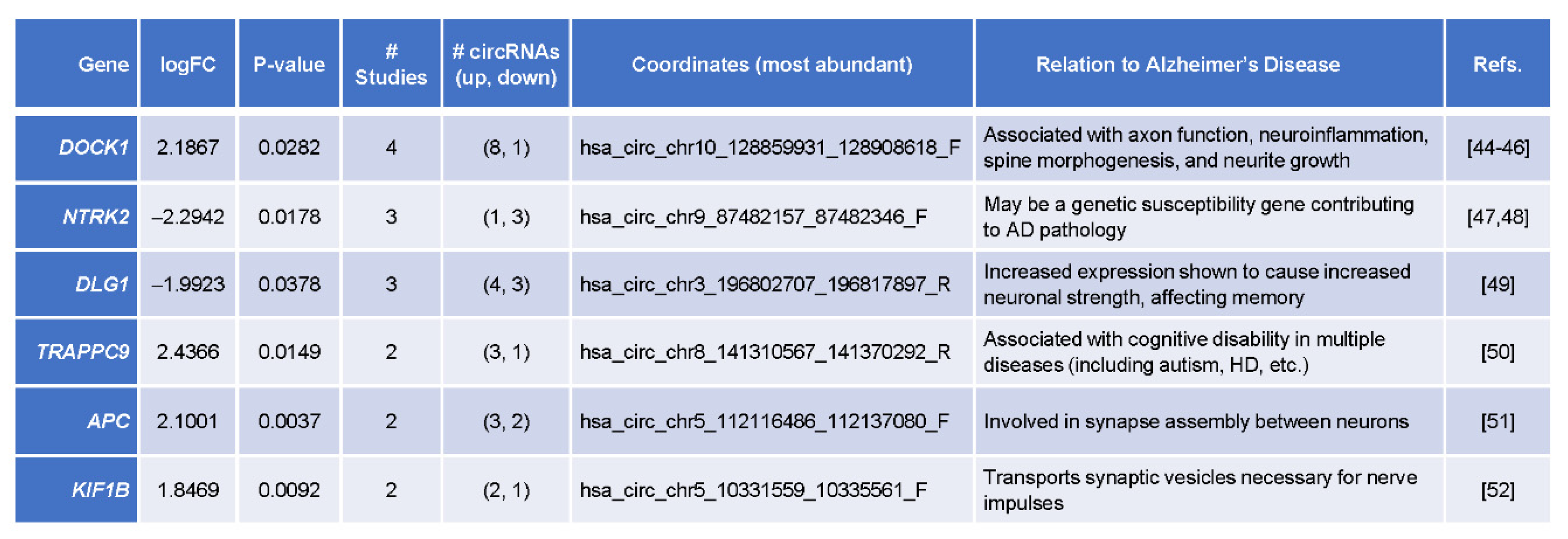
3.5. Top Differentially Abundant circRNAs Originated from Genes Implicated in AD Pathogenesis
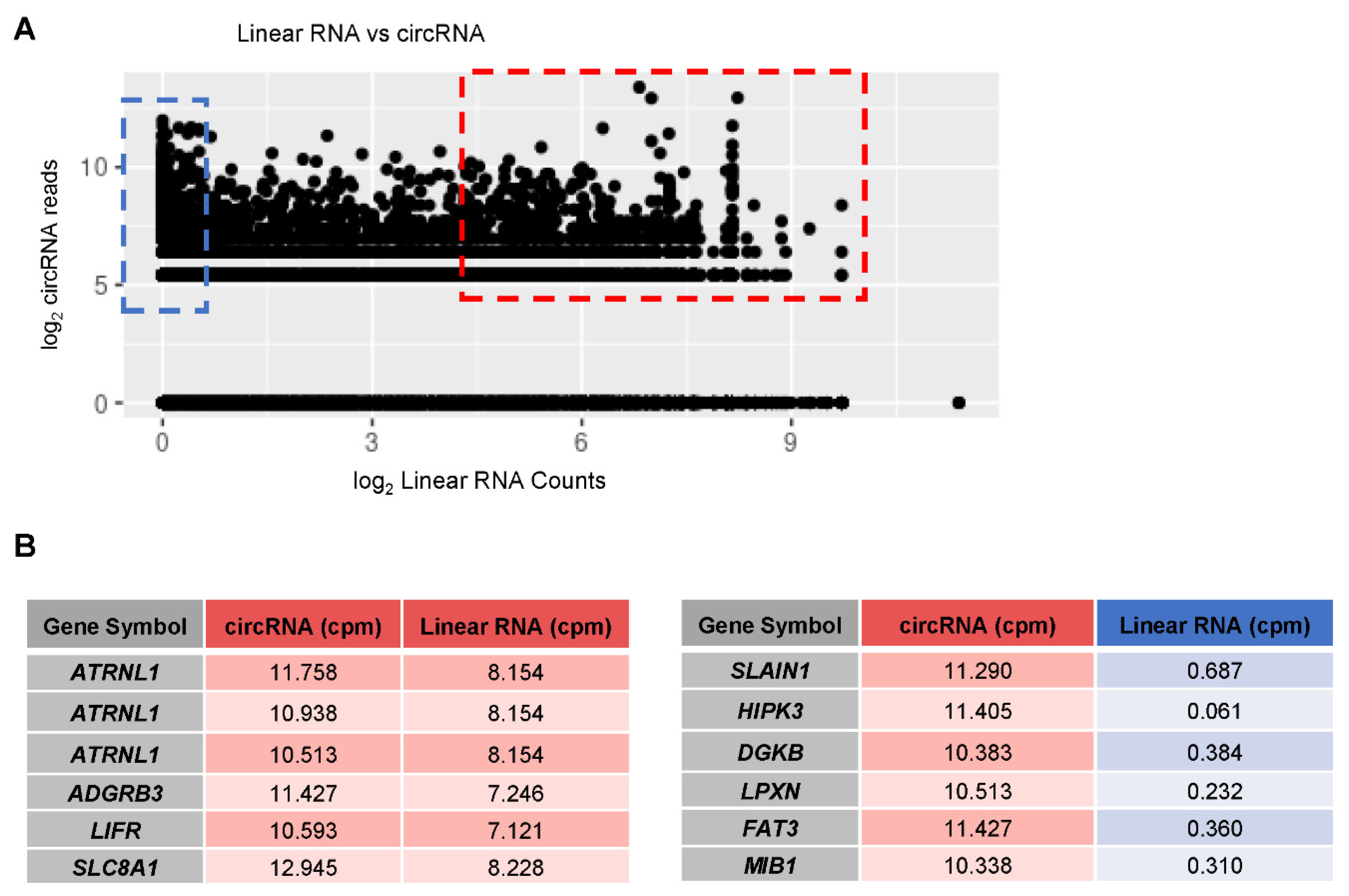
3.6. Linear vs. circRNA Expression in AD Samples
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xiao, M.S.; Ai, Y.; Wilusz, J.E. Biogenesis and Functions of Circular RNAs Come into Focus. Trends Cell Biol. 2020, 30, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yang, L.I.; Chen, L.-L. The Biogenesis, Functions, and Challenges of Circular RNAs. Mol. Cell 2018, 71, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panda, A.C.; De, S.; Grammatikakis, I.; Munk, R.; Yang, X.; Piao, Y.; Dudekula, D.B.; Abdelmohsen, K.; Gorospe, M. High-purity circular RNA isolation method (RPAD) reveals vast collection of intronic circRNAs. Nucleic Acids Res. 2017, 45, e116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.L. The expanding regulatory mechanisms and cellular functions of circular RNAs. Nat. Rev. Mol. Cell Biol. 2020, 21, 475–490. [Google Scholar] [CrossRef]
- Wilusz, J.E. A 360 degrees view of circular RNAs: From biogenesis to functions. Wiley Interdiscip. Rev. RNA 2018, 9, e1478. [Google Scholar] [CrossRef] [Green Version]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Rybak-Wolf, A.; Stottmeister, C.; Glažar, P.; Jens, M.; Pino, N.; Giusti, S.; Hanan, M.; Behm, M.; Bartok, O.; Ashwal-Fluss, R.; et al. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol. Cell 2015, 58, 870–885. [Google Scholar] [CrossRef] [Green Version]
- Panda, A.C.; Grammatikakis, I.; Munk, R.; Gorospe, M.; Abdelmohsen, K. Emerging roles and context of circular RNAs. Wiley Interdiscip. Rev. RNA 2017, 8, e1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Alexandrov, P.N.; Jaber, V.; Lukiw, W.J. Deficiency in the Ubiquitin Conjugating Enzyme UBE2A in Alzheimer’s Disease (AD) is Linked to Deficits in a Natural Circular miRNA-7 Sponge (circRNA; ciRS-7). Genes 2016, 7, 116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodbele, S.; Mutlu, N.; Wilusz, J.E. Best practices to ensure robust investigation of circular RNAs: Pitfalls and tips. EMBO Rep. 2021, 22, e52072. [Google Scholar] [CrossRef] [PubMed]
- Tsitsipatis, D.; Gorospe, M. Practical guide for circular RNA analysis: Steps, tips, and resources. Wiley Interdiscip. Rev. RNA 2021, 12, e1633. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, C.X.; Xue, W.; Zhang, Y.; Jiang, S.; Yin, Q.F.; Wei, J.; Yao, R.W.; Yang, L.; Chen, L.L. Coordinated circRNA biogenesis and function with NF90/NF110 in viral infection. Mol. Cell 2017, 67, 214–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdelmohsen, K.; Panda, A.C.; Munk, R.; Grammatikakis, I.; Dudekula, D.B.; De, S.; Kim, J.; Noh, J.H.; Kim, K.M.; Martindale, J.L.; et al. Identification of HuR target circular RNAs uncovers suppression of PABPN1 translation by CircPABPN1. RNA Biol. 2017, 14, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.R.; Yang, J.H.; Tsitsipatis, D.; Panda, A.C.; Noh, J.H.; Kim, K.M.; Munk, R.; Nicholson, T.; Hanniford, D.; Argibay, D.; et al. circSamd4 represses myogenic transcriptional activity of PUR proteins. Nucleic Acids Res. 2020, 155, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Ashwal-Fluss, R.; Meyer, M.; Pamudurti, N.R.; Ivanov, A.; Bartok, O.; Hanan, M.; Evantal, N.; Memczak, S.; Rajewsky, N.; Kadener, S. circRNA biogenesis competes with pre-mRNA splicing. Mol. Cell 2014, 56, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Du, W.W.; Yang, W.; Liu, E.; Yang, Z.; Dhaliwal, P.; Yang, B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016, 44, 2846–2858. [Google Scholar] [CrossRef] [Green Version]
- Legnini, I.; Di Timoteo, G.; Rossi, F.; Morlando, M.; Briganti, F.; Sthandier, O.; Fatica, A.; Santini, T.; Andronache, A.; Wade, M.; et al. Circ-ZNF609 is a circular RNA that can be translated and functions in myogenesis. Mol. Cell 2017, 66, 22–37. [Google Scholar] [CrossRef] [Green Version]
- Pamudurti, N.R.; Bartok, O.; Jens, M.; Ashwal-Fluss, R.; Stottmeister, C.; Ruhe, L.; Hanan, M.; Wyler, E.; Perez-Hernandez, D.; Ramberger, E.; et al. Translation of CircRNAs. Mol. Cell 2017, 66, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Weigelt, C.M.; Sehgal, R.; Tain, L.S.; Cheng, J.; Eßer, J.; Pahl, A.; Dieterich, C.; Grönke, S.; Partridge, L. An insulin-sensitive circular RNA that regulates lifespan in Drosophila. Mol. Cell 2020, 79, 268–279. [Google Scholar] [CrossRef]
- Stagsted, L.V.; Nielsen, K.M.; Daugaard, I.; Hansen, T.B. Noncoding AUG circRNAs constitute an abundant and conserved subclass of circles. Life Sci. Alliance 2019, 2, e201900398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verduci, L.; Tarcitano, E.; Strano, S.; Yarden, Y.; Blandino, G. CircRNAs: Role in human diseases and potential use as biomarkers. Cell Death Dis. 2021, 12, 468. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.L.; Dempsey, R.J.; Vemuganti, R. Role of circular RNAs in brain development and CNS diseases. Prog. Neurobiol. 2020, 186, 101746. [Google Scholar] [CrossRef]
- Jeck, W.R.; Sharpless, N.E. Detecting and characterizing circular RNAs. Nat. Biotechnol. 2014, 32, 453–461. [Google Scholar] [CrossRef]
- Verduci, L.; Strano, S.; Yarden, Y.; Blandino, G. The circRNA-microRNA code: Emerging implications for cancer diagnosis and treatment. Mol. Oncol. 2019, 13, 669–680. [Google Scholar] [CrossRef] [Green Version]
- Aufiero, S.; Reckman, Y.J.; Pinto, Y.M.; Creemers, E.E. Circular RNAs open a new chapter in cardiovascular biology. Nat. Rev. Cardiol. 2019, 16, 503–514. [Google Scholar] [CrossRef]
- Xu, H.; Guo, S.; Li, W.; Yu, P. The circular RNA Cdr1as, via miR-7 and its targets, regulates insulin transcription and secretion in islet cells. Sci. Rep. 2015, 5, 12453. [Google Scholar] [CrossRef]
- Scheckel, C.; Drapeau, E.; Frias, M.A.; Park, C.Y.; Fak, J.; Zucker-Scharff, I.; Kou, Y.; Haroutunian, V.; Ma’ayan, A.; Buxbaum, J.D.; et al. Regulatory consequences of neuronal ELAV-like protein binding to coding and non-coding RNAs in human brain. Elife 2016, 5, e10421. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Marshall, L.; Oh, G.; Jakubowski, J.L.; Groot, D.; He, Y.; Wang, T.; Petronis, A.; Labrie, V. Epigenetic dysregulation of enhancers in neurons is associated with Alzheimer’s disease pathology and cognitive symptoms. Nat. Commun. 2019, 10, 2246. [Google Scholar] [CrossRef] [Green Version]
- Magistri, M.; Velmeshev, D.; Makhmutova, M.; Faghihi, M.A. Transcriptomics Profiling of Alzheimer’s Disease Reveal Neurovascular Defects, Altered Amyloid-β Homeostasis, and Deregulated Expression of Long Noncoding RNAs. J. Alzheimers Dis. 2015, 48, 647–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nativio, R.; Donahue, G.; Berson, A.; Lan, Y.; Amlie-Wolf, A.; Tuzer, F.; Toledo, J.B.; Gosai, S.J.; Gregory, B.D.; Torres, C.; et al. Dysregulation of the epigenetic landscape of normal aging in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 497–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Pandini, C.; Bordoni, M.; Pansarasa, O.; Rey, F.; Costa, A.; Minafra, B.; Diamanti, L.; Zucca, S.; Carelli, S.; et al. Alzheimer’s, Parkinson’s Disease and Amyotrophic Lateral Sclerosis Gene Expression Patterns Divergence Reveals Different Grade of RNA Metabolism Involvement. Int. J. Mol. Sci. 2020, 21, 9500. [Google Scholar] [CrossRef] [PubMed]
- Toden, S.; Zhuang, J.; Acosta, A.; Karns, A.; Salathia, N.; Brewer, J.; Wilcock, D.; Aballi, J.; Nerenberg, M.; Quake, S.; et al. Noninvasive characterization of Alzheimer’s disease by circulating, cell-free messenger RNA next-generation sequencing. Sci. Adv. 2020, 6, 50. [Google Scholar] [CrossRef]
- Leinonen, R.; Sugawara, H.; Shumway, M. International Nucleotide Sequence Database Collaboration. The sequence read archive. Nucleic Acids Res. 2011, 39, D19–D50. [Google Scholar] [CrossRef] [Green Version]
- Hunt, S.E.; McLaren, W.; Gil, L.; Thormann, A.; Schuilenburg, H.; Sheppard, D.; Parton, A.; Armean, I.M.; Trevanion, S.J.; Flicek, P.; et al. Ensembl variation resources. Database 2018, 2018, bay119. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2: Accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, A.; Pimentel, H.; Trapnell, C.; Pachter, L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics 2011, 27, 2325–2329. [Google Scholar] [CrossRef]
- Barrett, S.P.; Salzman, J. Circular RNAs: Analysis, expression and potential functions. Development 2016, 143, 1838–1847. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019, 47, e47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stelzer, G.; Rosen, R.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Iny Stein, T.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analysis. Curr. Protoc. Bioinform. 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Yin, W.; Han, H.; Miller, H.; Li, J.; Herrada, A.A.; Kubo, M.; Sui, Z.; Gong, Q.; et al. The regulation of DOCK family proteins on T and B cells. J. Leukoc. Biol. 2021, 109, 383–394. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Gao, X.; Liu, G.; Xiong, W.; Wu, J.; Rao, Y. Netrin signal transduction and the guanine nucleotide exchange factor DOCK180 in attractive signaling. Nat. Neurosci. 2008, 11, 28–35. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Chung, J.; Panitch, R.; Zhu, C.; Beecham, G.W.; Mez, J.; Farrer, L.A.; Stein, T.D.; Crane, P.K.; Jun, G.R. Defining Alzheimer’s disease subtypes using polygenic risk scores integrated with genomic and brain transcriptomic profiles: Genetics/genetic factors of Alzheimer’s disease. Alzheimer’s Dement. 2020, 16, e046449. [Google Scholar] [CrossRef]
- Chen, Z.; Simmons, M.S.; Perry, R.T.; Wiener, H.W.; Harrell, L.E.; Go, R.C. Genetic association of neurotrophic tyrosine kinase receptor type 2 (NTRK2) With Alzheimer’s disease. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2008, 147, 363–369. [Google Scholar] [CrossRef]
- Zeng, F.; Zou, H.Q.; Zhou, H.D.; Li, J.; Wang, L.; Cao, H.Y.; Yi, X.; Wang, X.; Liang, C.R.; Wang, Y.R.; et al. The relationship between single nucleotide polymorphisms of the NTRK2 gene and sporadic Alzheimer’s disease in the Chinese Han population. Neurosci. Lett. 2013, 550, 55–59. [Google Scholar] [CrossRef]
- Walch, L. Emerging role of the scaffolding protein Dlg1 in vesicle trafficking. Traffic 2013, 14, 964–973. [Google Scholar] [CrossRef] [Green Version]
- Bodnar, B.; DeGruttola, A.; Zhu, Y.; Lin, Y.; Zhang, Y.; Mo, X.; Hu, W. Emerging role of NIK/IKK2-binding protein (NIBP)/trafficking protein particle complex 9 (TRAPPC9) in nervous system diseases. Transl. Res. 2020, 224, 55–70. [Google Scholar] [CrossRef]
- Fuchsberger, T.; Lloret, A.; Viña, J. New Functions of APC/C Ubiquitin Ligase in the Nervous System and Its Role in Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentil, B.J.; Cooper, L. Molecular basis of axonal dysfunction and traffic impairments in CMT. Brain Res. Bull. 2012, 88, 444–453. [Google Scholar] [CrossRef]
- Salzman, J.; Chen, R.E.; Olsen, M.N.; Wang, P.L.; Brown, P.O. Cell-type specific features of circular RNA expression. PLoS Genet. 2013, 9, e1003777. [Google Scholar] [CrossRef]
- Salzman, J.; Gawad, C.; Wang, P.L.; Lacayo, N.; Brown, P.O. Circular RNAs are the predominant transcript isoform from hundreds of human genes in diverse cell types. PLoS ONE 2012, 7, e30733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, K.M.; Meng, Q.; Perez de Acha, O.; Mustapic, M.; Cheng, A.; Eren, E.; Kundu, G.; Piao, Y.; Munk, R.; Wood, W.H., 3rd; et al. Mitochondrial RNA in Alzheimer’s Disease Circulating Extracellular Vesicles. Front Cell Dev. Biol. 2020, 8, 581882. [Google Scholar] [CrossRef]
- Dube, U.; Del-Aguila, J.L.; Li, Z.; Budde, J.P.; Jiang, S.; Hsu, S.; Ibanez, L.; Fernandez, M.V.; Farias, F.; Norton, J.; et al. An atlas of cortical circular RNA expression in Alzheimer disease brains demonstrates clinical and pathological associations. Nat. Neurosci. 2019, 22, 1903–1912. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cochran, K.R.; Veeraraghavan, K.; Kundu, G.; Mazan-Mamczarz, K.; Coletta, C.; Thambisetty, M.; Gorospe, M.; De, S. Systematic Identification of circRNAs in Alzheimer’s Disease. Genes 2021, 12, 1258. https://doi.org/10.3390/genes12081258
Cochran KR, Veeraraghavan K, Kundu G, Mazan-Mamczarz K, Coletta C, Thambisetty M, Gorospe M, De S. Systematic Identification of circRNAs in Alzheimer’s Disease. Genes. 2021; 12(8):1258. https://doi.org/10.3390/genes12081258
Chicago/Turabian StyleCochran, Kyle R., Kirtana Veeraraghavan, Gautam Kundu, Krystyna Mazan-Mamczarz, Christopher Coletta, Madhav Thambisetty, Myriam Gorospe, and Supriyo De. 2021. "Systematic Identification of circRNAs in Alzheimer’s Disease" Genes 12, no. 8: 1258. https://doi.org/10.3390/genes12081258
APA StyleCochran, K. R., Veeraraghavan, K., Kundu, G., Mazan-Mamczarz, K., Coletta, C., Thambisetty, M., Gorospe, M., & De, S. (2021). Systematic Identification of circRNAs in Alzheimer’s Disease. Genes, 12(8), 1258. https://doi.org/10.3390/genes12081258