
Transcriptomic Changes Following Partial Depletion of CENP-E in Normal Human Fibroblasts

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Culture Conditions
2.2. RNA Interference
2.3. Reverse Transcription Quantitative PCR (RT-qPCR)
2.4. Microarray and Bioinformatics Analysis
2.5. Gene Set Enrichment Analysis (GSEA)
3. Results
3.1. Transcriptome and Bioinformatics Analyses
3.2. Validation of Arrays Data Using RT-qPCR Analysis
3.3. Gene Set Enrichment Analysis (GSEA)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gordon, D.J.; Resio, B.; Pellman, D. Causes and Consequences of Aneuploidy in Cancer. Nat. Rev. Genet. 2012, 13, 189–203. [Google Scholar] [CrossRef]
- Thompson, S.L.; Bakhoum, S.F.; Compton, D.A. Mechanisms of Chromosomal Instability. Curr. Biol. CB 2010, 20, R285–R295. [Google Scholar] [CrossRef] [Green Version]
- Thompson, S.L.; Compton, D.A. Chromosomes and Cancer Cells. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2011, 19, 433–444. [Google Scholar] [CrossRef] [Green Version]
- Owa, M.; Dynlacht, B. A Non-Canonical Function for Centromere-Associated Protein-E Controls Centrosome Integrity and Orientation of Cell Division. Commun. Biol. 2021, 4, 358. [Google Scholar] [CrossRef]
- Yen, T.J.; Li, G.; Schaar, B.T.; Szilak, I.; Cleveland, D.W. CENP-E Is a Putative Kinetochore Motor That Accumulates Just before Mitosis. Nature 1992, 359, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Silk, A.D.; Zasadil, L.M.; Holland, A.J.; Vitre, B.; Cleveland, D.W.; Weaver, B.A. Chromosome Missegregation Rate Predicts Whether Aneuploidy Will Promote or Suppress Tumors. Proc. Natl. Acad. Sci. USA 2013, 110, E4134–E4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, B.A.A.; Silk, A.D.; Montagna, C.; Verdier-Pinard, P.; Cleveland, D.W. Aneuploidy Acts Both Oncogenically and as a Tumor Suppressor. Cancer Cell 2007, 11, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, B.A.; Cleveland, D.W. The Aneuploidy Paradox in Cell Growth and Tumorigenesis. Cancer Cell 2008, 14, 431–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneziano, L.; Barra, V.; Cilluffo, D.; Di Leonardo, A. Proliferation of Aneuploid Cells Induced by CENP-E Depletion Is Counteracted by the P14ARF Tumor Suppressor. Mol. Genet. Genom. MGG 2019, 294, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, R.; Gonzalez-Angulo, A.-M.; Myhre, S.; Carey, M.; Lee, J.-S.; Overgaard, J.; Alsner, J.; Stemke-Hale, K.; Lluch, A.; Neve, R.M.; et al. Integrative Analysis of Cyclin Protein Levels Identifies Cyclin B1 as a Classifier and Predictor of Outcomes in Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 3654–3662. [Google Scholar] [CrossRef] [Green Version]
- Yen, T.J.; Compton, D.A.; Wise, D.; Zinkowski, R.P.; Brinkley, B.R.; Earnshaw, W.C.; Cleveland, D.W. CENP-E, a Novel Human Centromere-Associated Protein Required for Progression from Metaphase to Anaphase. EMBO J. 1991, 10, 1245–1254. [Google Scholar] [CrossRef]
- Mao, Y.; Desai, A.; Cleveland, D.W. Microtubule Capture by CENP-E Silences BubR1-Dependent Mitotic Checkpoint Signaling. J. Cell Biol. 2005, 170, 873–880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Legal, T.; Hayward, D.; Gluszek-Kustusz, A.; Blackburn, E.A.; Spanos, C.; Rappsilber, J.; Gruneberg, U.; Welburn, J.P.I. The C-Terminal Helix of BubR1 Is Essential for CENP-E-Dependent Chromosome Alignment. J. Cell Sci. 2020, 133, jcs246025. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Holland, A.J.; Lan, W.; Cleveland, D.W. Aurora Kinases and Protein Phosphatase 1 Mediate Chromosome Congression through Regulation of CENP-E. Cell 2010, 142, 444–455. [Google Scholar] [CrossRef] [Green Version]
- El-Arabey, A.A.; Salama, S.A.; Abd-Allah, A.R. CENP-E as a Target for Cancer Therapy: Where Are We Now? Life Sci. 2018, 208, 192–200. [Google Scholar] [CrossRef]
- Cilluffo, D.; Barra, V.; Spatafora, S.; Coronnello, C.; Contino, F.; Bivona, S.; Feo, S.; Di Leonardo, A. Aneuploid IMR90 Cells Induced by Depletion of PRB, DNMT1 and MAD2 Show a Common Gene Expression Signature. Genomics 2020, 112, 2541–2549. [Google Scholar] [CrossRef]
- Fragkos, M.; Barra, V.; Egger, T.; Bordignon, B.; Lemacon, D.; Naim, V.; Coquelle, A. Dicer Prevents Genome Instability in Response to Replication Stress. Oncotarget 2019, 10, 4407–4423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oncomine Login. Available online: https://www.oncomine.org//resource/login.html (accessed on 21 August 2021).
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [Green Version]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- GSEA. Available online: https://www.gsea-msigdb.org/gsea/index.jsp (accessed on 20 August 2021).
- Bennett, A.; Bechi, B.; Tighe, A.; Thompson, S.; Procter, D.J.; Taylor, S.S. Cenp-E Inhibitor GSK923295: Novel Synthetic Route and Use as a Tool to Generate Aneuploidy. Oncotarget 2015, 6, 20921–20932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ciossani, G.; Overlack, K.; Petrovic, A.; Huis In’t Veld, P.J.; Koerner, C.; Wohlgemuth, S.; Maffini, S.; Musacchio, A. The Kinetochore Proteins CENP-E and CENP-F Directly and Specifically Interact with Distinct BUB Mitotic Checkpoint Ser/Thr Kinases. J. Biol. Chem. 2018, 293, 10084–10101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, P.; Miotto, B.; Saint-Ruf, C.; Said, M.; Barra, V.; Nähse, V.; Ravera, S.; Cappelli, E.; Naim, V. FANCD2 Modulates the Mitochondrial Stress Response to Prevent Common Fragile Site Instability. Commun. Biol. 2021, 4, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New Insights into the Mechanisms of Epithelial-Mesenchymal Transition and Implications for Cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
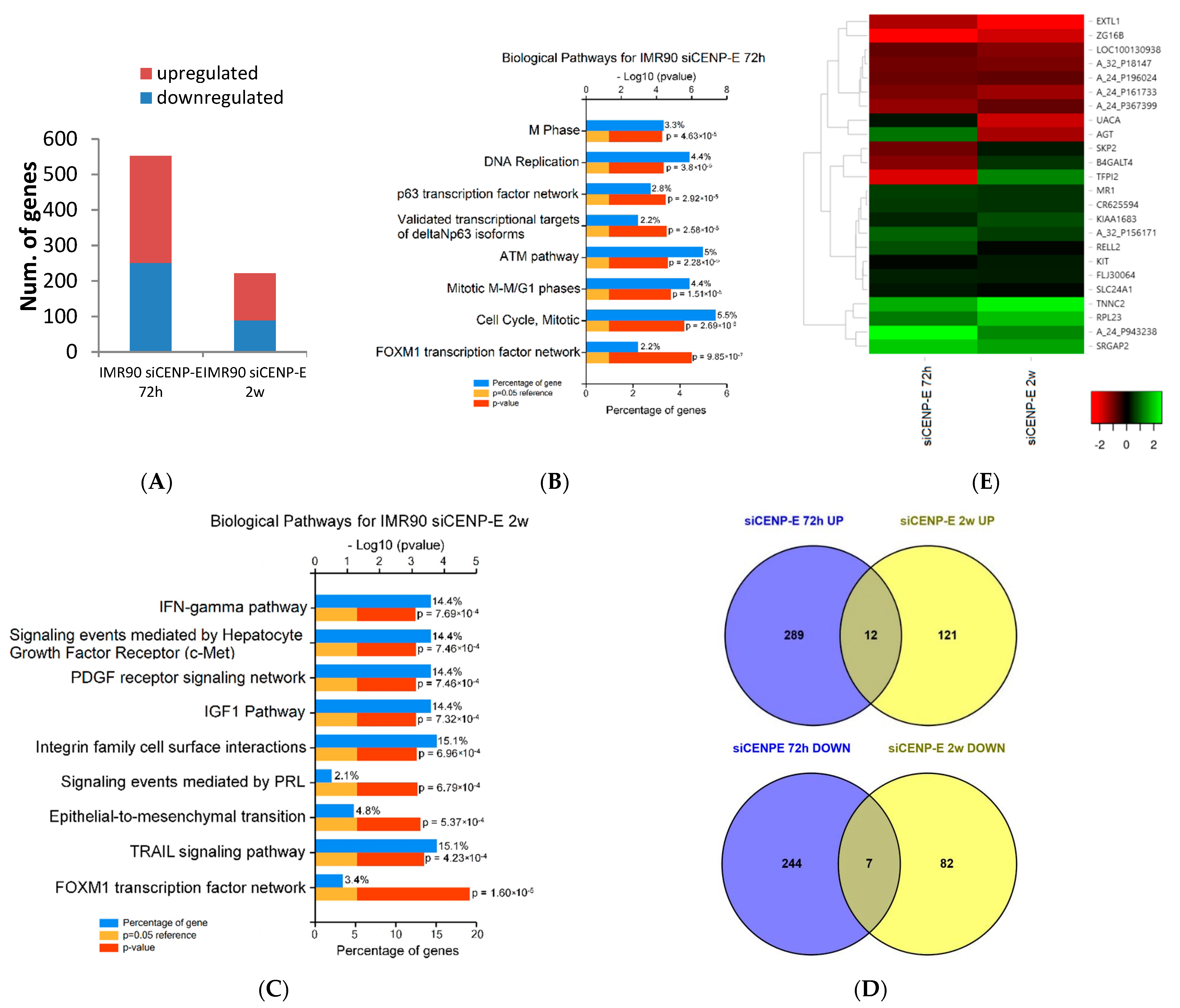
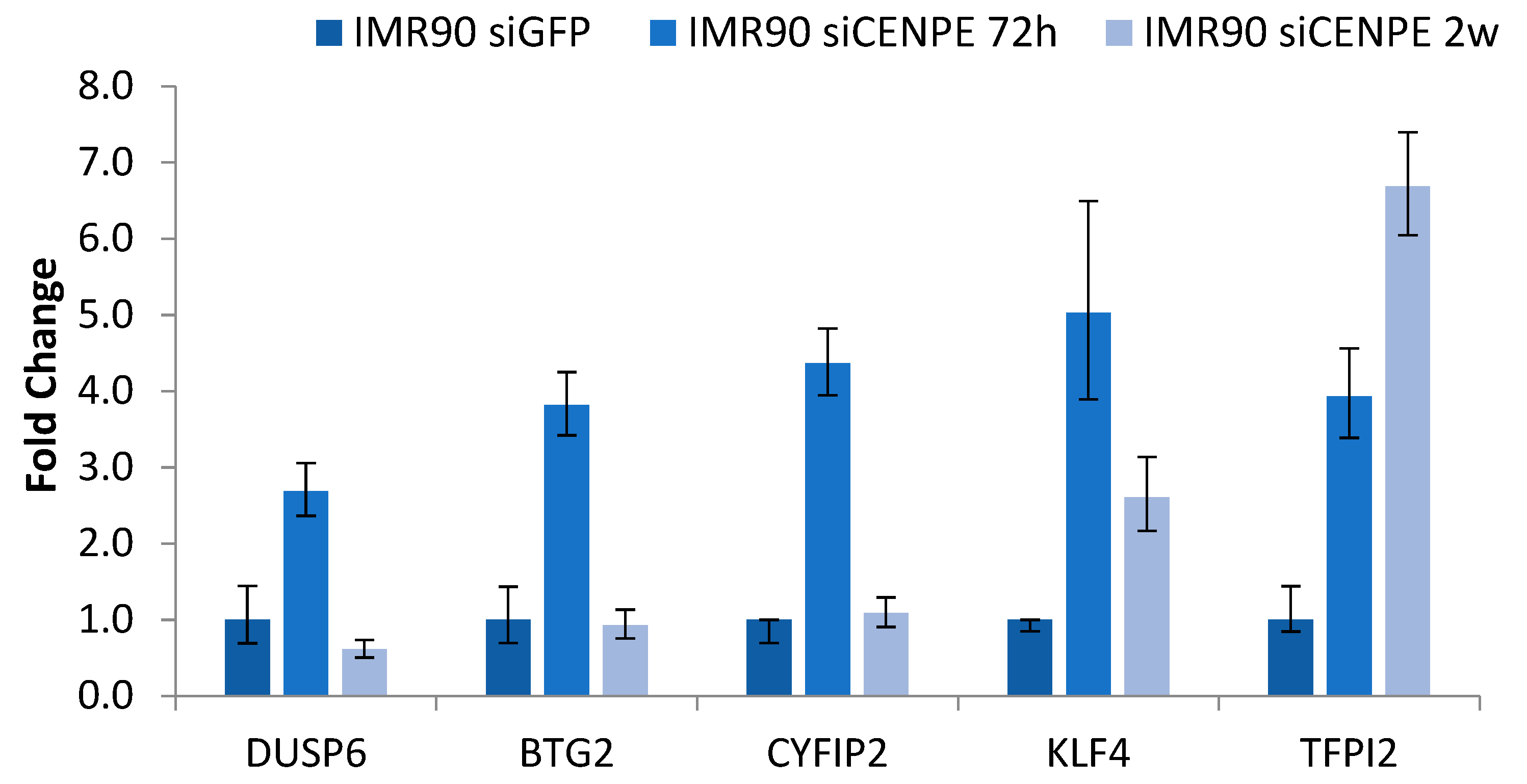
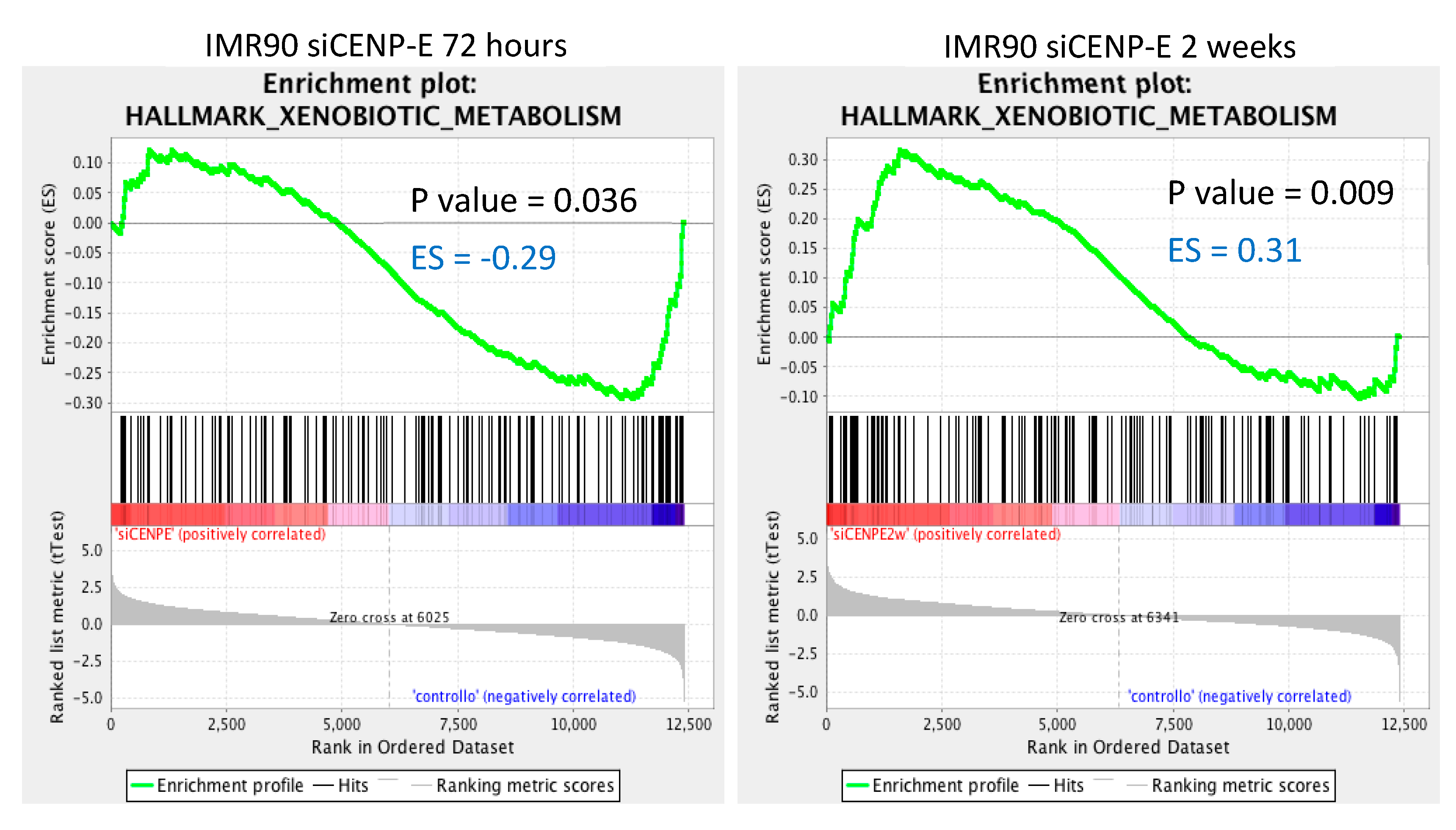

{kind=link}
{kind=link}
{kind=link}
| Gene ID | Description | LogFC | # Chr. | Map Location | |
|---|---|---|---|---|---|
| 72 h | 2 Weeks | ||||
| ZG16B | zymogen granule protein 16B | −2.21378 | −1.58077 | 16 | 16p13.3 |
| TFPI2 | tissue factor pathway inhibitor 2 | −1.73241 | 1.967329 | 7 | 7q22 |
| EXTL1 | Exostosin-like glycosyltransferase 1 | −1.27449 | −2.37647 | 1 | 1p36.1 |
| MR1 | major histocompatibility complex, class I-related | 1.173685 | 1.064675 | 1 | 1q25.3 |
| AGT | angiotensinogen (serpin peptidase inhibitor, clade A, member 8) | 1.752677 | −1.15911 | 1 | 1q42.2 |
| RPL23 | ribosomal protein L23 | 1.819593 | 2.630793 | 17 | 17q |
| TNNC2 | troponin C type 2 (fast) | 2.405029 | 3.232683 | 20 | 20q12-q13.11 |
| SRGAP2 | SLIT-ROBO Rho GTPase activating protein 2 | 2.733915 | 2.305187 | 1 | 1q32.1 |
| B4GALT4 | UDP-Gal:betaGlcNAc beta 1,4- galactosyltransferase, polypeptide 4 | −0.88068 | 1.067247 | 3 | 3q13.3 |
| UACA | uveal autoantigen with coiled-coil domains and ankyrin repeats | 0.669221 | −1.5223 | 15 | 15q22-q24 |
| KIAA1683 | Q Domain-Containing Protein N | 0.875501 | 1.339538 | 19 | 19p13.1 |
| SKP2 | S-phase kinase−associated protein 2, E3 ubiquitin protein ligase | −0.68236 | 0.753601 | 5 | 5p13 |
| KIT | v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog | 0.552419 | 0.785465 | 4 | 4q12 |
| RELL2 | RELT-like 2 | 1.337275 | 0.531247 | 5 | 5q31.3 |
| SLC24A1 | solute carrier family 24 (sodium/potassium/calcium exchanger), member 1 | 0.701308 | 0.546544 | 15 | 15q22 |
| Hallmark Gene set 72 h | SIZE | ES | NES | p-Value | FDR q-Value |
|---|---|---|---|---|---|
| Upregulated | |||||
| Cholesterol Homeostasis | 55 | 0.550825 | 2.219.529 | 0.0 | 0.0 |
| Apoptosis | 128 | 0.29306307 | 14.145.108 | 0.026666667 | 0.17741449 |
| Downregulated | |||||
| E2F Targets | 124 | −0.4112242 | −18.818.628 | 0 | 0.001760578 |
| G2/M Checkpoint | 130 | −0.42952272 | −19.638.364 | 0 | 0.001968572 |
| Mitotic Spindle | 132 | −0.33654705 | −15.422.429 | 0 | 0.08193885 |
| Angiogenesis | 25 | −0.44820115 | −14.857.963 | 0.04587156 | 0.100587666 |
| Estrogen Response Late | 160 | −0.30400178 | −14.472.796 | 0.013745705 | 0.11437272 |
| Bile Acid Metabolism | 81 | −0.33262056 | −14.141.632 | 0.028520498 | 0.12994246 |
| Xenobiotic Metabolism | 145 | −0.29437807 | −13.890.945 | 0.036036037 | 0.14013477 |
| Unfolded Protein Response | 74 | −0.31752646 | −13.405.416 | 0.060998153 | 0.14493392 |
| Adipogenesis | 130 | −0.29467914 | −1.363.687 | 0.03345725 | 0.15041217 |
| TNFA Signaling via NFkB | 142 | −0.28777105 | −13.413.501 | 0.041736227 | 0.15983883 |
| Hallmark Gene set 2 weeks | SIZE | ES | NES | p-vauel | FDR q-value |
| Upregulated | |||||
| Epithelial Mesenchymal Transition | 151 | 0.33510843 | 1.519.574 | 0.0075642965 | 0.11385157 |
| IL6/JAK/STAT Signaling | 58 | 0.37629685 | 14.682.375 | 0.034035657 | 0.11548446 |
| Interferone Gamma Response | 129 | 0.33727473 | 14.752.915 | 0.012403101 | 0.1355435 |
| Hypoxia | 141 | 0.34279573 | 15.343.299 | 0.00147929 | 0.14622506 |
| Xenobiotic Metabolism | 145 | 0.31554386 | 14.156.777 | 0.009036144 | 0.15259206 |
| Coagulation | 100 | 0.32489774 | 13.754.646 | 0.032357473 | 0.16717246 |
| Heme Metabolism | 136 | 0.30528104 | 13.594.577 | 0.03153153 | 0.16950405 |
| Complement | 141 | 0.31029537 | 13.893.101 | 0.02413273 | 0.16991296 |
| Hedgeog Signaling | 24 | 0.41114244 | 13.165.202 | 0.12477396 | 0.17681527 |
| Inflammatory Response | 138 | 0.30268276 | 13.413.032 | 0.052469134 | 0.17737256 |
| UV Response DN | 104 | 0.3095823 | 13.233.161 | 0.046178345 | 0.18309012 |
| Downregulated | |||||
| mTORC1 Signaling | 141 | −0.33381698 | −16.185.987 | 0.0 | 0.047431484 |
| P53 Pathway | 135 | −0.34690675 | −16.548.364 | 0.005830904 | 0.06875795 |
| NOTCH Signaling | 24 | −0.45094362 | −14.955.359 | 0.04481132 | 0.09119969 |
| KRAS Signaling UP | 143 | −0.29774994 | −14.312.139 | 0.018018018 | 0.1097472 |
| Gene Symbol | Description | Running ES |
|---|---|---|
| TIAM1 | T-cell lymphoma invasion and metastasis 1 | −0.32938716 |
| LRPPRC | Leucine-rich PPR-motif-containing | −0.32254452 |
| FSCN1 | fascin homolog 1, actin-bundling protein (Strongylocentrotus purpuratus) | −0.31544223 |
| HDAC6 | histone deacetylase 6 | −0.31205016 |
| UXT | Ubiquitously-expressed transcript | −0.3070534 |
| TUBD1 | tubulin, delta 1 | −0.30694872 |
| CKAP5 | Cytoskeleton-associated protein 5 | −0.3005278 |
| TTK | TTK protein kinase | −0.2928824 |
| ARHGAP4 | Rho GTPase-activating protein 4 | −0.29161006 |
| MARCKS | myristoylated alanine-rich protein kinase C substrate | −0.29939783 |
| GEMIN4 | gem (nuclear organelle)-associated protein 4 | −0.29208082 |
| FBXO5 | F-box protein 5 | −0.2945973 |
| ROCK1 | Rho-associated, coiled-coil-containing protein kinase 1 | −0.29402748 |
| LLGL1 | lethal giant larvae homolog 1 (Drosophila) | −0.29998654 |
| PXN | Paxillin | −0.30580252 |
| CENPF | centromere protein F, 350/400 ka (mitosin) | −0.29683083 |
| PREX1 | Phosphatidylinositol 3,4,5-Trisphosphate-Dependent Rac Exchanger 1 Protein | −0.29543936 |
| RAB3GAP1 | RAB3 GTPase-activating protein subunit 1 (catalytic) | −0.28851372 |
| CENPJ | centromere protein J | −0.28630605 |
| KIF2C | kinesin family member 2C | −0.27653277 |
| TPX2 | TPX2, microtubule-associated, homolog (Xenopus laevis) | −0.26817042 |
| TSC1 | tuberous sclerosis 1 | −0.2579142 |
| NCK1 | NCK adaptor protein 1 | −0.25523746 |
| ARHGEF3 | Rho guanine nucleotide exchange factor (GEF) 3 | −0.2437383 |
| SYNPO | Synaptopodin | −0.2319448 |
| STK38L | Serine–threonine kinase 38-like | −0.22208665 |
| OPHN1 | oligophrenin 1 | −0.21298712 |
| BUB1 | BUB1 budding uninhibited by benzimidazoles 1 homolog (yeast) | −0.20986164 |
| KIF4A | kinesin family member 4° | −0.20093325 |
| ARHGAP29 | Rho GTPase-activating protein 29 | −0.18930057 |
| PRC1 | protein regulator of cytokinesis 1 | −0.17876585 |
| CLASP1 | Cytoplasmic-linker-associated protein 1 | −0.1655671 |
| TOP2A | topoisomerase (DNA) II alpha 170kDa | −0.16242237 |
| RHOF | ras homolog gene family, member F (in filopodia) | −0.14529541 |
| KIF15 | kinesin family member 15 | −0.12773909 |
| PCGF5 | polycomb group ring finger 5 | −0.11064719 |
| NEK2 | NIMA (never in mitosis gene a)-related kinase 2 | −0.09230707 |
| CCNB2 | cyclin B2 | −0.076223895 |
| KIF23 | kinesin family member 23 | −0.058513902 |
| RACGAP1 | Rac GTPase-activating protein 1 | −0.03790126 |
| CENPE | centromere protein E, 312 kDa | −0.018124685 |
| KIF11 | kinesin family member 11 | 0.005052448 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cilluffo, D.; Chiavetta, R.F.; Bivona, S.; Contino, F.; Coronnello, C.; Feo, S.; Di Leonardo, A.; Barra, V. Transcriptomic Changes Following Partial Depletion of CENP-E in Normal Human Fibroblasts. Genes 2021, 12, 1322. https://doi.org/10.3390/genes12091322
Cilluffo D, Chiavetta RF, Bivona S, Contino F, Coronnello C, Feo S, Di Leonardo A, Barra V. Transcriptomic Changes Following Partial Depletion of CENP-E in Normal Human Fibroblasts. Genes. 2021; 12(9):1322. https://doi.org/10.3390/genes12091322
Chicago/Turabian StyleCilluffo, Danilo, Roberta Flavia Chiavetta, Serena Bivona, Flavia Contino, Claudia Coronnello, Salvatore Feo, Aldo Di Leonardo, and Viviana Barra. 2021. "Transcriptomic Changes Following Partial Depletion of CENP-E in Normal Human Fibroblasts" Genes 12, no. 9: 1322. https://doi.org/10.3390/genes12091322
APA StyleCilluffo, D., Chiavetta, R. F., Bivona, S., Contino, F., Coronnello, C., Feo, S., Di Leonardo, A., & Barra, V. (2021). Transcriptomic Changes Following Partial Depletion of CENP-E in Normal Human Fibroblasts. Genes, 12(9), 1322. https://doi.org/10.3390/genes12091322