
Identification of CSF3R Mutations in B-Lineage Acute Lymphoblastic Leukemia Using Comprehensive Cancer Panel and Next-Generation Sequencing

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Karyotyping and FISH
2.3. DNA Isolation and Target Sequencing
2.4. Comprehensive Cancer Panel
2.5. Sequence Alignment and Variant Calling
2.6. Variant Annotation
2.7. Exploration of B-ALL Mutation Data Using Maftools
3. Results
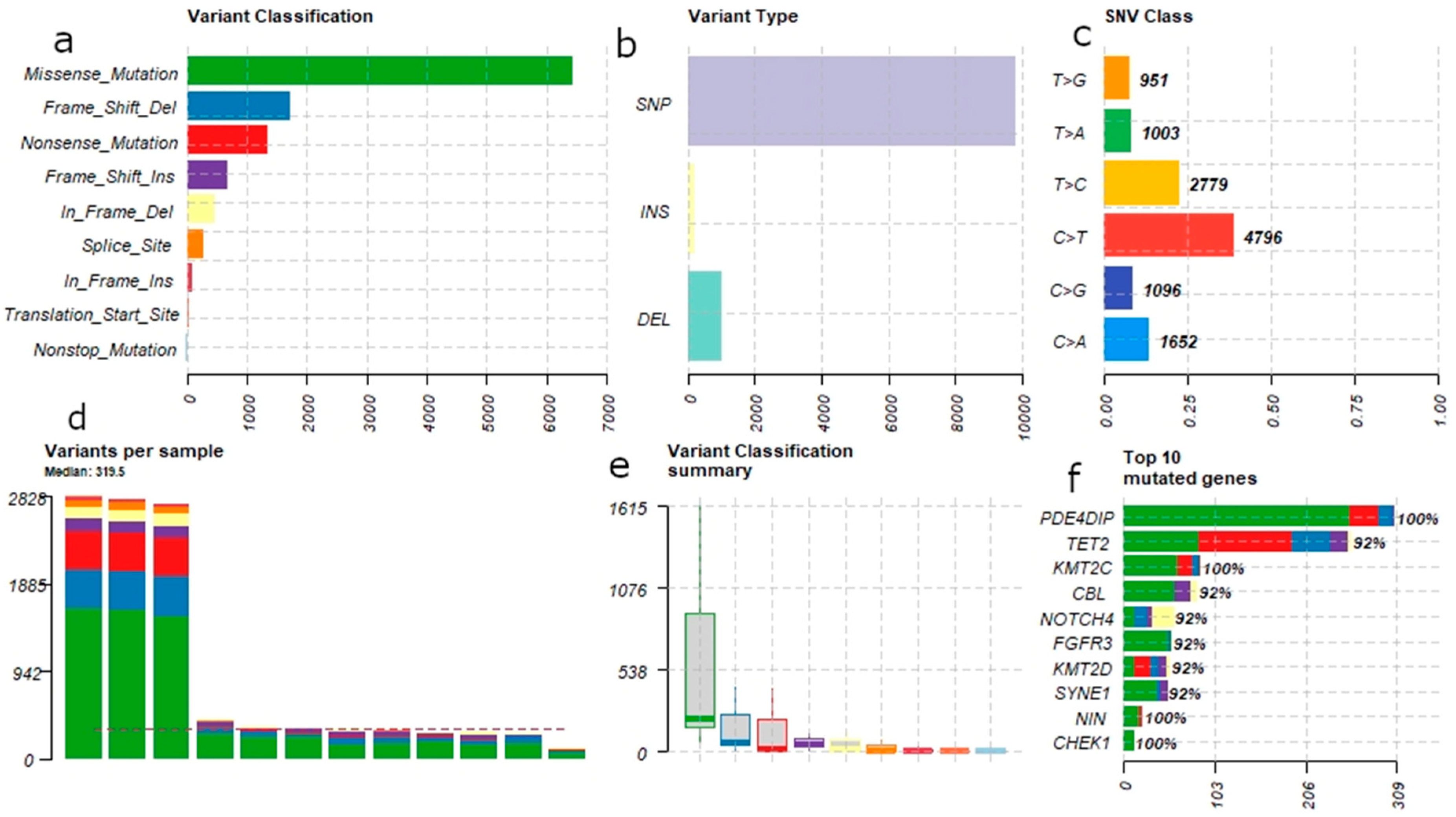
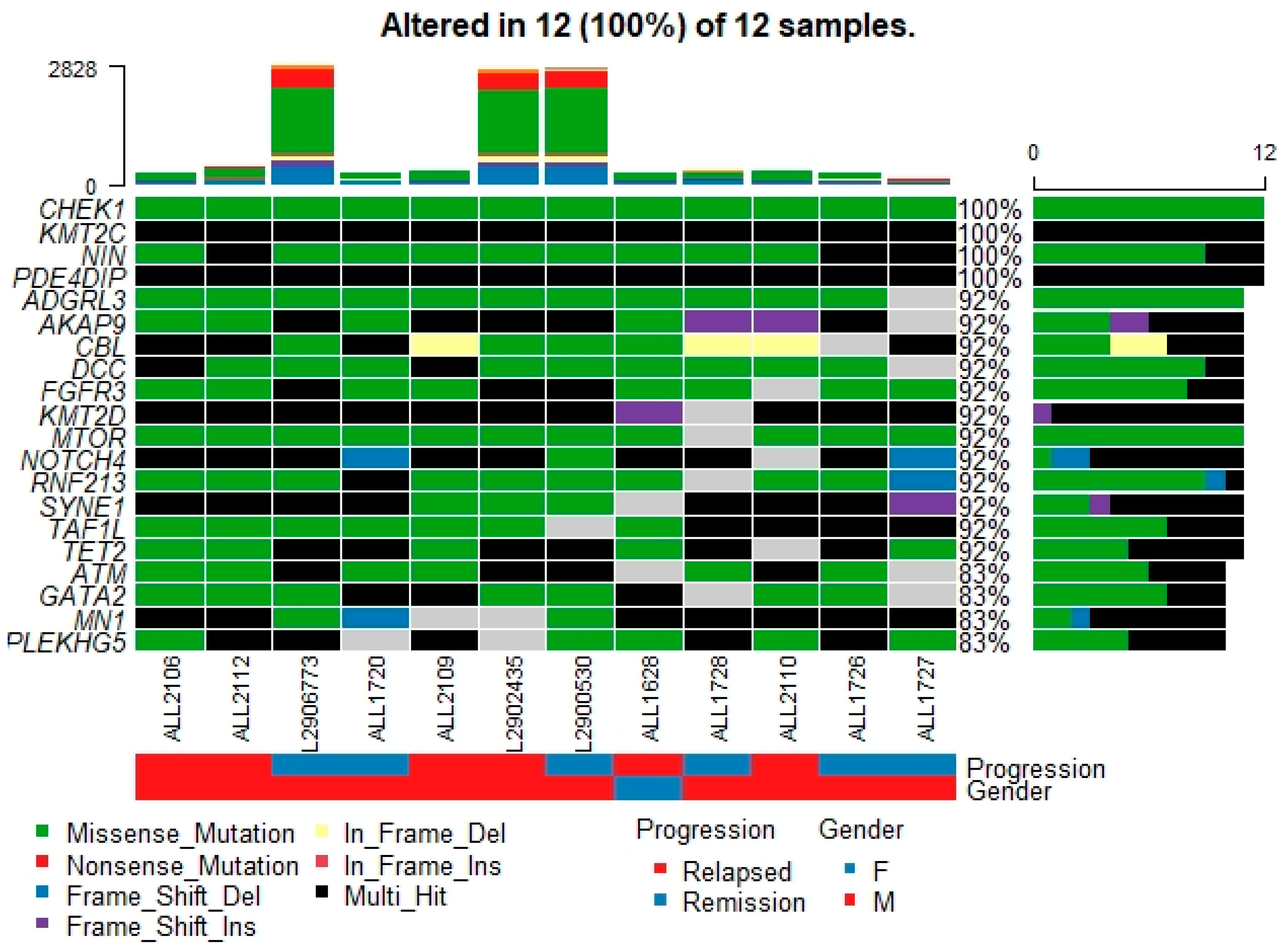
3.1. Mutational Landscape of B-ALL
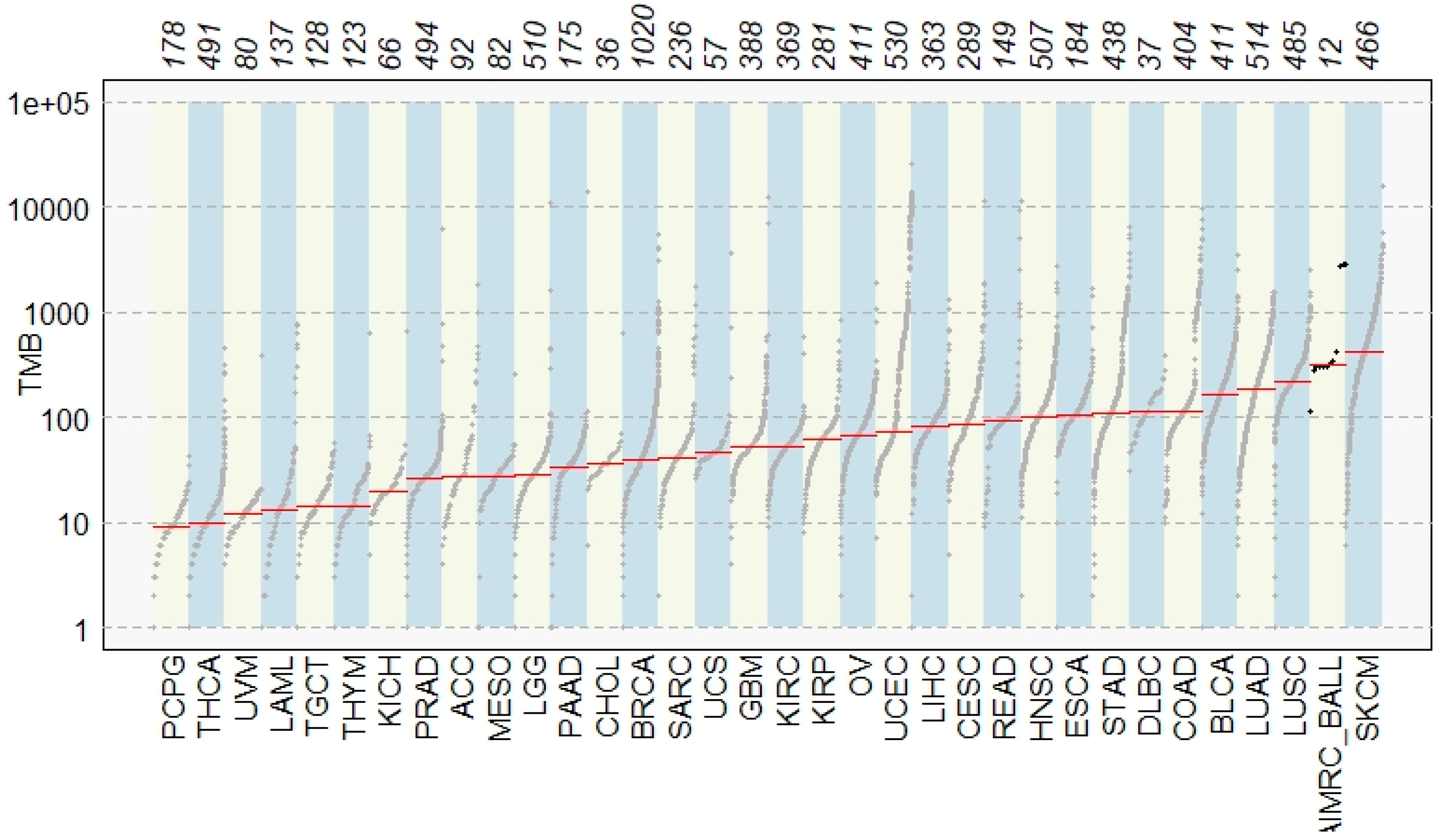
3.2. Comparison with TCGA Signature
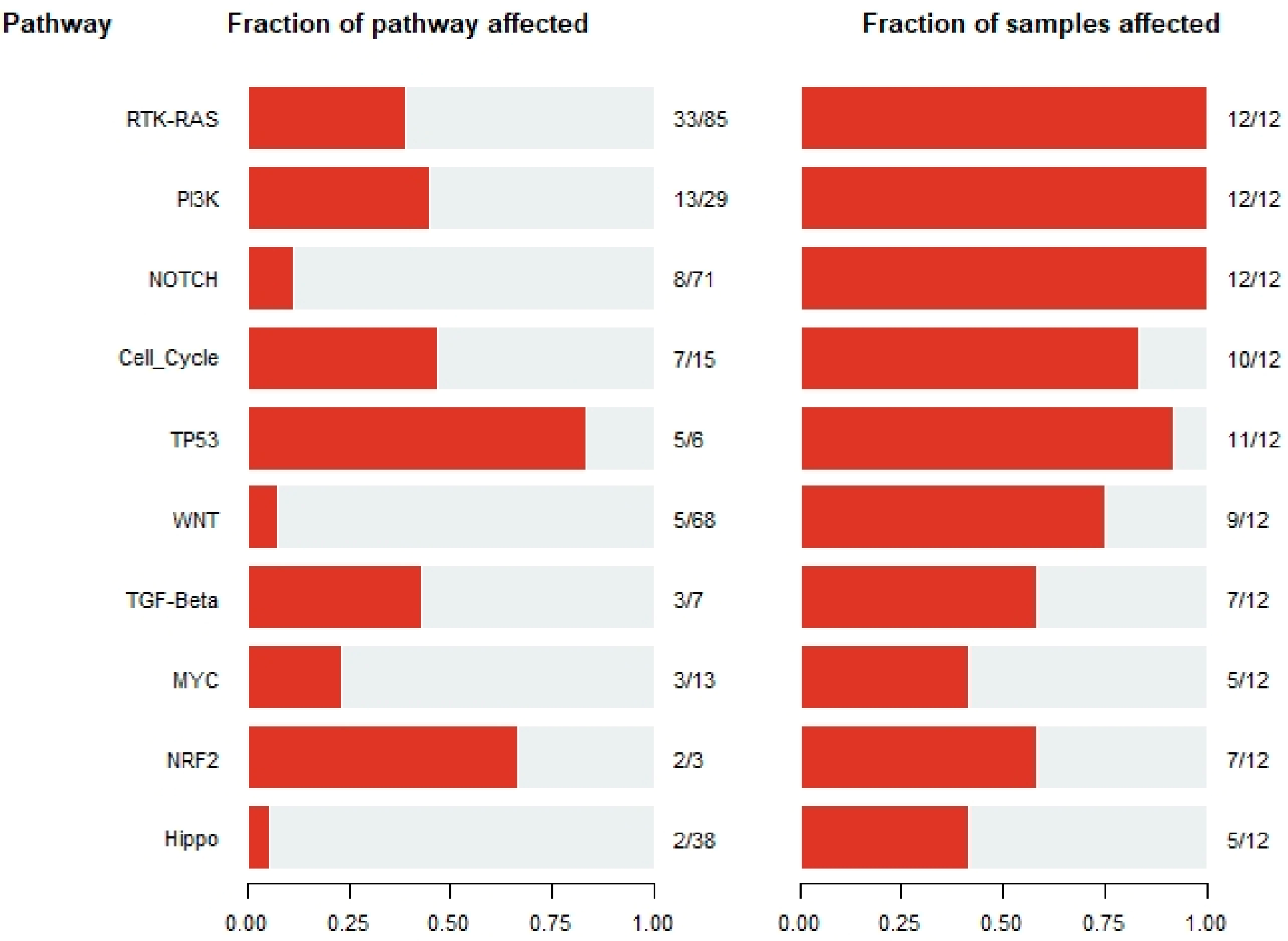
3.3. Signature Pathways across B-ALL Samples
3.4. Somatic Interaction of Mutations
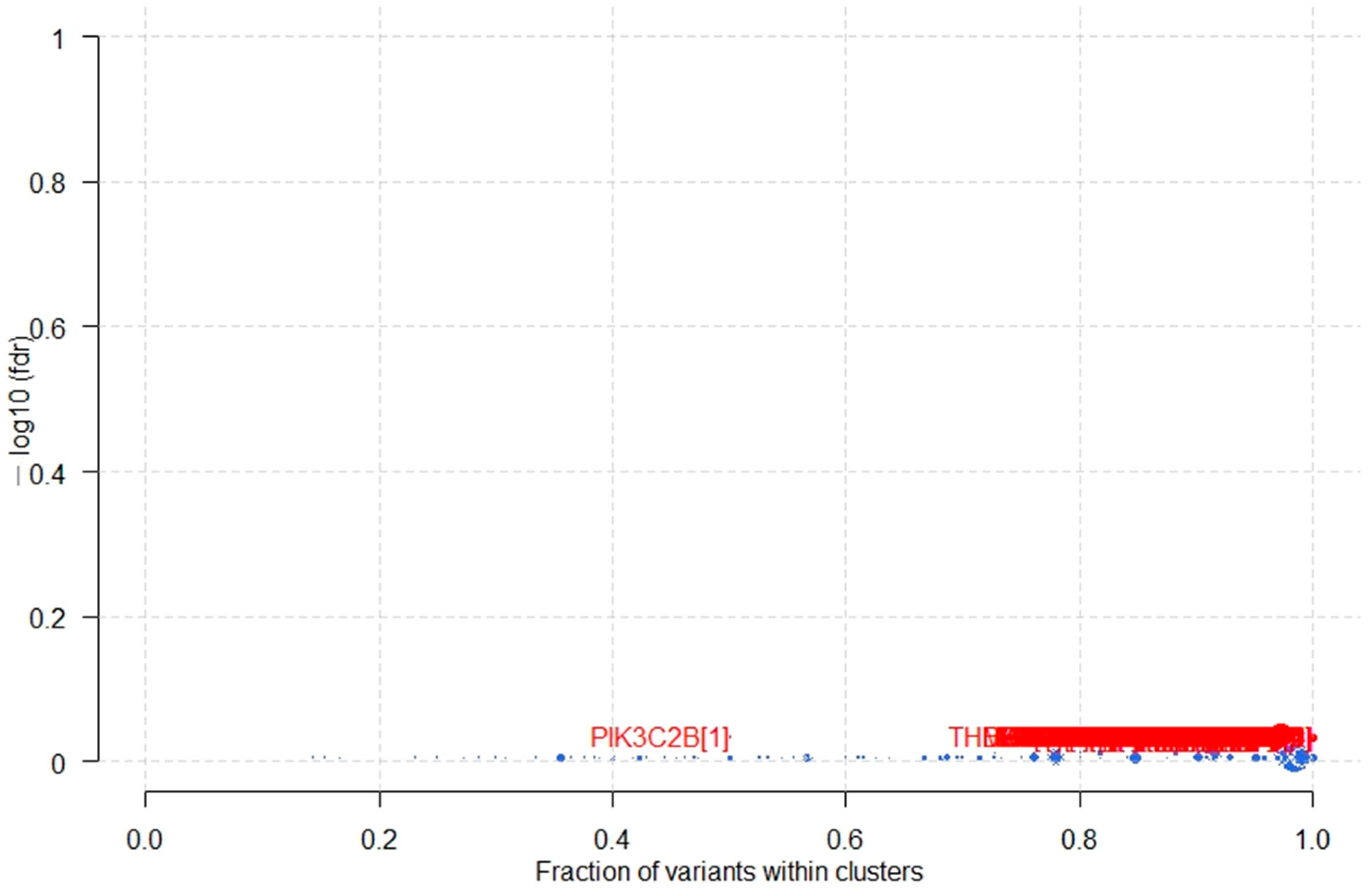
3.5. Potential Cancer Driver Genes in B-ALL
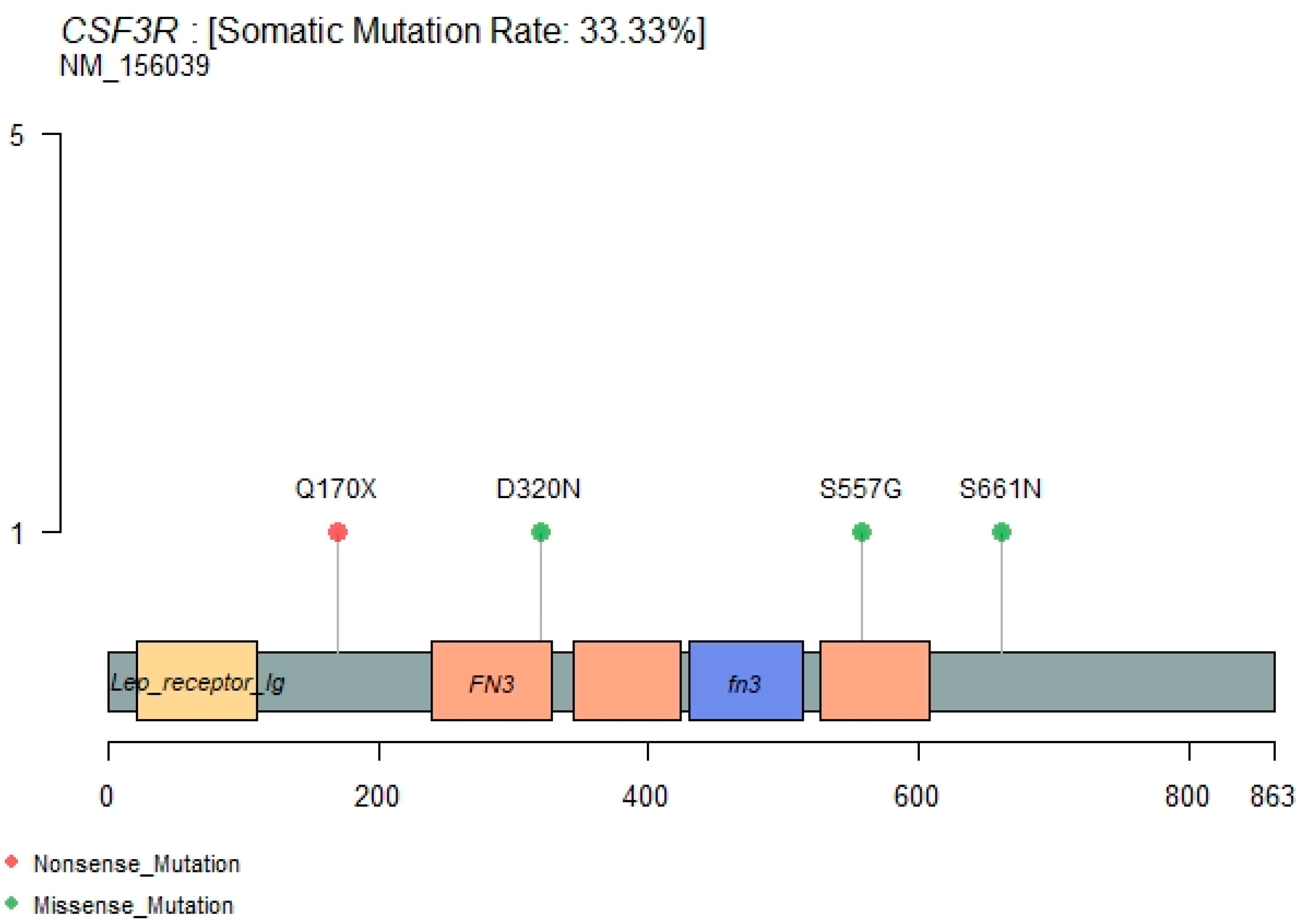
3.6. CSF3R Mutations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhou, Y.; Kanagal-Shamanna, R.; Zuo, Z.; Tang, G.; Medeiros, L.J.; Bueso-Ramos, C.E. Advances in B-lymphoblastic leukemia: Cytogenetic and genomic lesions. Ann. Diagn. Pathol. 2016, 23, 43–50. Available online: https://pubmed.ncbi.nlm.nih.gov/27130145/ (accessed on 21 August 2021). [CrossRef] [PubMed]
- Heerema, N.A.; Sather, H.N.; Sensel, M.G.; Liu-Mares, W.; Lange, B.J.; Bostrom, B.C.; Nachman, J.B.; Steinherz, P.G.; Hutchinson, R.; Gaynon, P.S.; et al. Association of chromosome arm 9p abnormalities with adverse risk in childhood acute lymphoblastic leukemia: A report from the Children’s Cancer Group. Blood 1999, 94, 1537–1544. Available online: http://ashpublications.org/blood/article-pdf/94/5/1537/1657032/1537.pdf (accessed on 21 August 2021). [PubMed]
- Meijerink, J.P.P.; den Boer, M.L.; Pieters, R. New Genetic Abnormalities and Treatment Response in Acute Lymphoblastic Leukemia. Semin. Hematol. 2009, 46, 16–23. Available online: https://pubmed.ncbi.nlm.nih.gov/19100364/ (accessed on 21 August 2021). [CrossRef] [PubMed]
- Xu, N.; Li, Y.L.; Zhou, X.; Cao, R.; Li, H.; Lu, Q.S.; Li, L.; Lu, Z.; Huang, J.; Sun, J.; et al. CDKN2 gene deletion as poor prognosis predictor involved in the progression of adult B-lineage acute lymphoblastic leukemia patients. J. Cancer 2015, 6, 1114–1120. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4615347/ (accessed on 21 August 2021). [CrossRef] [PubMed] [Green Version]
- Davidsson, J.; Paulsson, K.; Lindgren, D.; Lilljebjörn, H.; Chaplin, T.; Forestier, E.; Andersen, M.K.; Nordgren, A.; Rosenquist, R.; Fioretos, T.; et al. Relapsed childhood high hyperdiploid acute lymphoblastic leukemia: Presence of preleukemic ancestral clones and the secondary nature of microdeletions and RTK-RAS mutations. Leukemia 2010, 24, 924–931. Available online: http://projects.tcag.ca/variation/ (accessed on 21 August 2021). [CrossRef] [PubMed] [Green Version]
- Moorman, A.V. New and emerging prognostic and predictive genetic biomarkers in B-cell precursor acute lymphoblastic leukemia. Haematologica 2016, 101, 407–416. Available online: www.haematologica.org/content/101/4/407 (accessed on 21 August 2021). [CrossRef] [Green Version]
- Pendergrass, T.W. Epidemiology of acute lymphoblastic leukemia. Semin. Oncol. 1985, 12, 80–91. Available online: https://europepmc.org/article/med/3859924 (accessed on 21 August 2021). [PubMed]
- Mullighan, C.G. The molecular genetic makeup of acute lymphoblastic leukemia. [Internet]. Hematol. Am. Soc. Hematol. Educ. Progr. 2012, 2012, 389–396. Available online: https://pubmed.ncbi.nlm.nih.gov/23233609/ (accessed on 21 August 2021). [CrossRef] [Green Version]
- Tasian, S.K.; Loh, M.L.; Hunger, S.P. Childhood acute lymphoblastic leukemia: Integrating genomics into therapy. Cancer 2015, 121, 3577–3590. Available online: https://pubmed.ncbi.nlm.nih.gov/26194091/ (accessed on 21 August 2021). [CrossRef]
- Pui, C.H.; Nichols, K.E.; Yang, J.J. Somatic and germline genomics in paediatric acute lymphoblastic leukaemia. Nat. Rev. Clin. Oncol. 2019, 16, 227–240. Available online: https://pubmed.ncbi.nlm.nih.gov/30546053/ (accessed on 21 August 2021). [CrossRef] [PubMed]
- Nahi, H.; Hägglund, H.; Ahlgren, T.; Bernell, P.; Hardling, M.; Karlsson, K.; Lazarevic, V.L.; Linderholm, M.; Smedmyr, B.; Aström, M.; et al. An investigation into whether deletions in 9p reflect prognosis in adult precursor B-cell acute lymphoblastic leukemia: A multi-center study of 381 patients. Haematologica 2008, 93, 1734–1738. Available online: https://pubmed.ncbi.nlm.nih.gov/18728022/ (accessed on 21 August 2021). [CrossRef] [Green Version]
- Chessells, J.M.; Harrison, G.; Lilleyman, J.S.; Bailey, C.C.; Richards, S.M. Continuing (maintenance) therapy in lymphoblastic leukaemia: Lessons from MRC UKALL X. Br. J. Haematol. 1997, 98, 945–951. Available online: https://pubmed.ncbi.nlm.nih.gov/9326194/ (accessed on 21 August 2021). [CrossRef]
- Moorman, A.V.; Harrison, C.J.; Buck, G.A.N.; Richards, S.M.; Secker-Walker, L.M.; Martineau, M.; Vance, G.H.; Cherry, A.M.; Higgins, R.R.; Fielding, A.K.; et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): Analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood 2007, 109, 3189–3197. Available online: https://pubmed.ncbi.nlm.nih.gov/17170120/ (accessed on 21 August 2021). [CrossRef] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genom. Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Wang, H.; Qian, X.; Gao, S.; Xia, J.; Liu, J.; Cheng, Y.; Man, J.; Zhai, X. Genetic mutational analysis of pediatric acute lymphoblastic leukemia from a single center in China using exon sequencing. BMC Cancer 2020, 20, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. Available online: https://pubmed.ncbi.nlm.nih.gov/23884480/ (accessed on 21 August 2021). [CrossRef] [PubMed]
- Tarlock, K.; Alonzo, T.; Wang, Y.C.; Gerbing, R.B.; Ries, R.E.; Hylkema, T.; Smith, J.L.; Maxson, J.E.; Meshinchi, S. Prognostic impact of CSF3R mutations in favorable risk childhood acute myeloid leukemia. Blood 2020, 135, 1603–1606. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, F.; Chen, X.; Zhang, Y.; Wang, M.; Liu, H.; Cao, P.; Ma, X.; Wang, X.; Zhang, J.; et al. CSF3R Mutations are frequently associated with abnormalities of RUNX1, CBFB, CEBPA, and NPM1 genes in acute myeloid leukemia. Cancer 2018, 124, 3329–3338. Available online: https://pubmed.ncbi.nlm.nih.gov/29932212/ (accessed on 21 August 2021). [CrossRef] [Green Version]
- Schwartz, M.S.; Wieduwilt, M.J. CSF3R truncation mutations in a patient with B-cell acute lymphoblastic leukemia and a favorable response to chemotherapy plus dasatinib. Leuk. Res. Rep. 2020, 14, 100208. [Google Scholar] [PubMed]
- Maxson, J.E.; Gotlib, J.; Pollyea, D.A.; Fleischman, A.G.; Agarwal, A.; Eide, C.A.; Bottomly, D.; Wilmot, B.; McWeeney, S.K.; Tognon, C.E.; et al. Oncogenic CSF3R mutations in chronic neutrophilic leukemia atypical. N. Engl. J. Med. 2013, 368, 1781–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Age at Diagnosis (Years)/Sex | WBC | Blast | Cytogenetic | FISH | Progression |
|---|---|---|---|---|---|---|
| 1628 | 4/F | 27.1 | 32% | 46,XX,i(22)(?q10)(3)/46,XX(19) | nuc ish(P16x0,9cenx2), (BCRx4,ABL1x2)(61/200)/(BCRx3,ABL1x2)(37/200), (P53x2,D17Z1x3)(34/200)/(P53,D17Z1)x3(20/200) | Relapse |
| 1720 | 6/M | 1.4 | 60% | NA | Nuc ish(p16x0,9cenx2,D17Z1x3)(17/200) | Remission |
| 1726 | 12/M | 6.3 | 30% | 44-46,XY,del(2)(p?22),?dic(9;17)(p?13;p?11.2),?der(14;17)(?p11.2;?p13),+mar,inc(cp3) | 9p deletion | Remission |
| 1727 | 3/M | 10.5 | 85% | 46,XY,del(11)(q13q23),inc(19)/46,XY (2) | 9p, MLL deletion and TEL/AML fusion | Remission |
| 1728 | 8/M | 9.5 | 13% | 52-55,XY,+X,+X,+4,+6,+8,dic(9;17)t(?p22;q10),+14,+18,+21,+21,inc(cp12)/46,XY(5) | 9p deletion and hyperdiploidy | Remission |
| 2106 * | 8/M | N.P | 80% | 46,XY(18) | NA | Relapsed |
| 2109 | 8/M | 5.2 | 3% | 46,XY,del(9)(p21)(8)/46,XY(12) | 9p deletion | Relapsed |
| 2110 * | 10/M | 7.9 | 80% | 46,XY(20) | TEL/AML positive | Relapsed |
| 2112 * | 3/M | 16.5 | 84% | 46,XY(20) | Negative | Relapsed |
| 2902435 | 11/M | 1.09 | 4% | 46,XY(18) | 9p21,extra copy of aml1 | Relapsed |
| 2906773 | 15/M | 10 | 19% | 46,XY,del(9)(q21),del(13)(q21)(17) | P16 gene deletion | Remission |
| 2900530 | 20/M | 2.8 | 30% | NA | P16 gene deletion | Remission |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rashid, M.; Alasiri, A.; Al Balwi, M.A.; Alkhaldi, A.; Alsuhaibani, A.; Alsultan, A.; Alharbi, T.; Alomair, L.; Almuzzaini, B. Identification of CSF3R Mutations in B-Lineage Acute Lymphoblastic Leukemia Using Comprehensive Cancer Panel and Next-Generation Sequencing. Genes 2021, 12, 1326. https://doi.org/10.3390/genes12091326
Rashid M, Alasiri A, Al Balwi MA, Alkhaldi A, Alsuhaibani A, Alsultan A, Alharbi T, Alomair L, Almuzzaini B. Identification of CSF3R Mutations in B-Lineage Acute Lymphoblastic Leukemia Using Comprehensive Cancer Panel and Next-Generation Sequencing. Genes. 2021; 12(9):1326. https://doi.org/10.3390/genes12091326
Chicago/Turabian StyleRashid, Mamoon, Abdulrahman Alasiri, Mohammad A. Al Balwi, Aziza Alkhaldi, Ahmed Alsuhaibani, Abdulrahman Alsultan, Talal Alharbi, Lamya Alomair, and Bader Almuzzaini. 2021. "Identification of CSF3R Mutations in B-Lineage Acute Lymphoblastic Leukemia Using Comprehensive Cancer Panel and Next-Generation Sequencing" Genes 12, no. 9: 1326. https://doi.org/10.3390/genes12091326
APA StyleRashid, M., Alasiri, A., Al Balwi, M. A., Alkhaldi, A., Alsuhaibani, A., Alsultan, A., Alharbi, T., Alomair, L., & Almuzzaini, B. (2021). Identification of CSF3R Mutations in B-Lineage Acute Lymphoblastic Leukemia Using Comprehensive Cancer Panel and Next-Generation Sequencing. Genes, 12(9), 1326. https://doi.org/10.3390/genes12091326