
Gene Duplication and Gene Fusion Are Important Drivers of Tumourigenesis during Cancer Evolution


Abstract
1. Introduction
2. Routes to Gene Duplication in Cancer
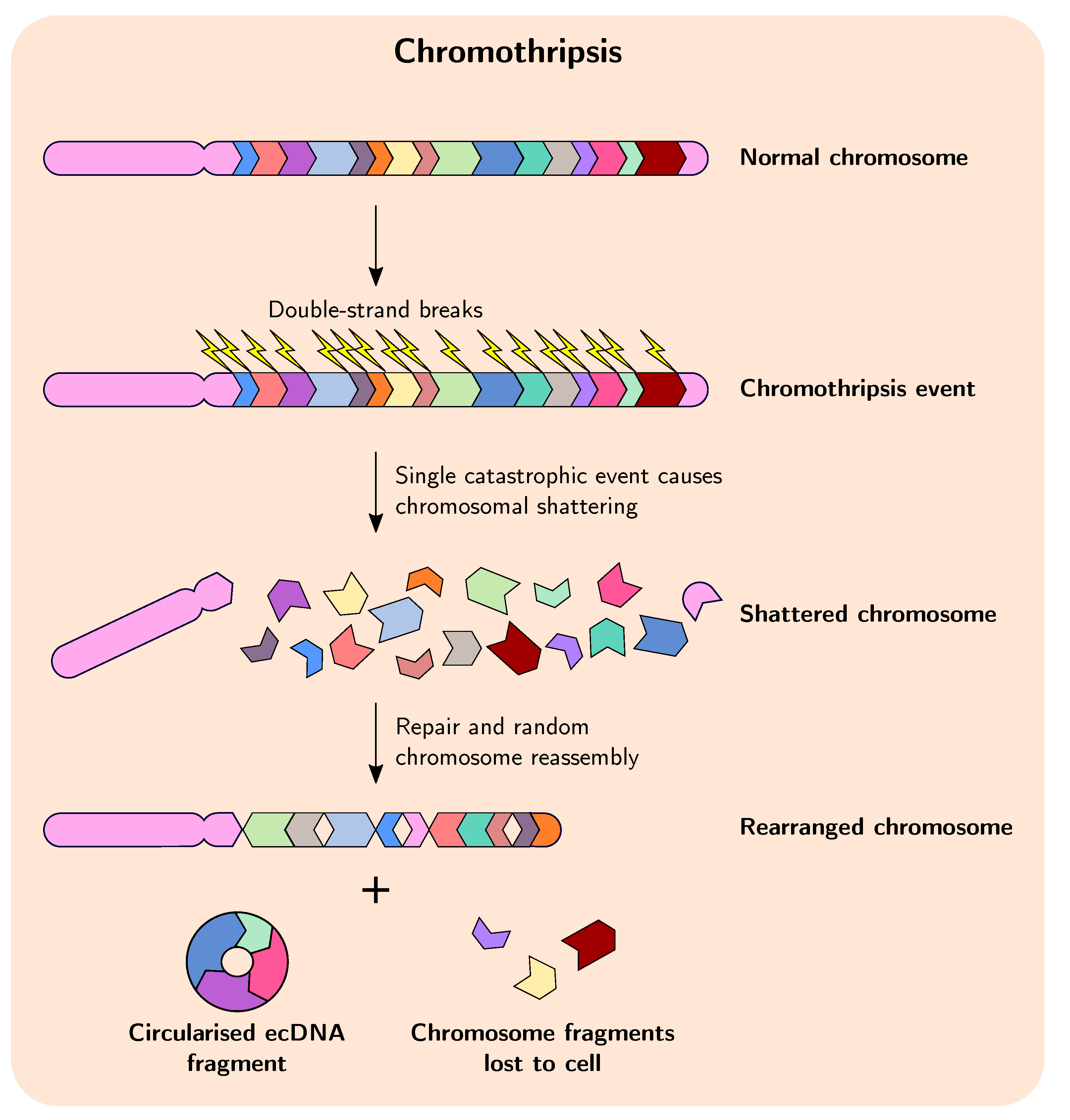
2.1. Chromothripsis
2.2. Whole-Genome and Whole-Chromosome Duplication
3. Frequency of Structural Variation Leading to Duplication in Cancer
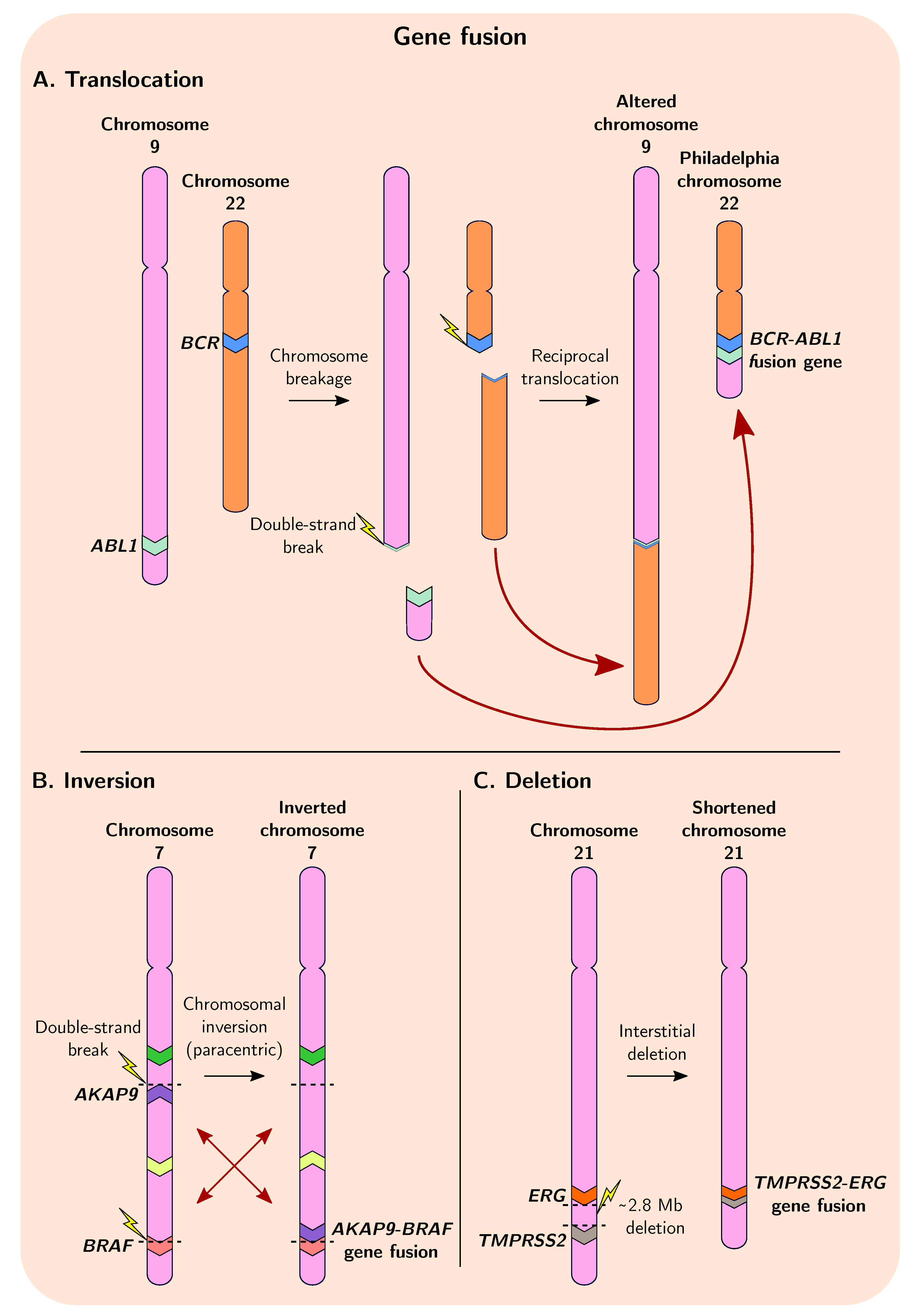
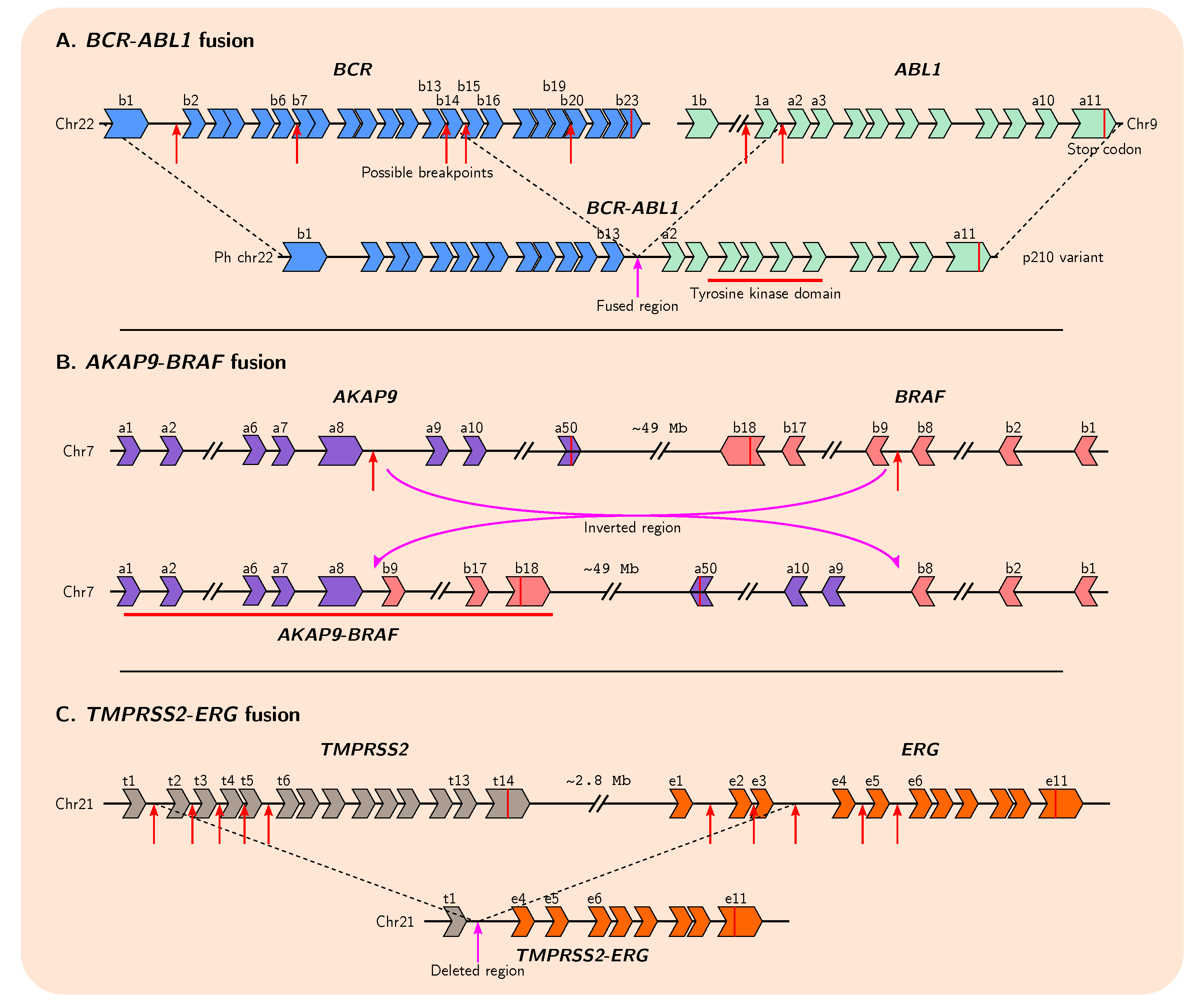
4. Gene Fusions Give Rise to Genetic Novelty


4.1. Tools for Detection of Fusion Genes
4.2. Role of Fusion Genes in Cancer Evolution
4.3. Therapeutic Relevance of Fusion Genes
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DHFR | Dihydrofolate reductase |
| ecDNA | Extrachromosomal DNA |
| DM | Double minute |
| WGD | Whole-genome duplication |
| WCD | Whole-chromosome duplication |
| CIN | Chromosomal instability |
| PCAWG | Pan-cancer analysis of whole genomes |
| SCNA | Somatic copy number alteration |
| CML | Chronic myeloid leukaemia |
| RNA-seq | RNA sequencing |
| MDCAGFC | Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer |
| RTK | Receptor tyrosine kinase |
References
- Stephens, S. Possible significance of duplication in evolution. Adv. Genet. 1951, 4, 247–265. [Google Scholar]
- Nei, M. Gene duplication and nucleotide substitution in evolution. Nature 1969, 221, 40–42. [Google Scholar] [CrossRef]
- Ohno, S. Evolution by Gene Duplication; Springer: Berlin/Heidelberg, Germany, 1970. [Google Scholar]
- Conant, G.C.; Wolfe, K.H. Turning a hobby into a job: How duplicated genes find new functions. Nat. Rev. Genet. 2008, 9, 938–950. [Google Scholar] [CrossRef]
- Innan, H.; Kondrashov, F. The evolution of gene duplications: Classifying and distinguishing between models. Nat. Rev. Genet. 2010, 11, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA: Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Li, Y.; Roberts, N.D.; Wala, J.A.; Shapira, O.; Schumacher, S.E.; Kumar, K.; Khurana, E.; Waszak, S.; Korbel, J.O.; Haber, J.E.; et al. Patterns of somatic structural variation in human cancer genomes. Nature 2020, 578, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Solomon, E.; Borrow, J.; Goddard, A. Chromosome aberrations and cancer. Science 1991, 254, 1153–1160. [Google Scholar] [CrossRef]
- Storchova, Z.; Pellman, D. From polyploidy to aneuploidy, genome instability and cancer. Nat. Rev. Mol. Cell Biol. 2004, 5, 45–54. [Google Scholar] [CrossRef]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Long, M.; Langley, C.H. Natural selection and the origin of jingwei, a chimeric processed functional gene in Drosophila. Science 1993, 260, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Mitelman, F.; Johansson, B.; Mertens, F. The impact of translocations and gene fusions on cancer causation. Nat. Rev. Cancer 2007, 7, 233–245. [Google Scholar] [CrossRef]
- Fisher, J.C. Multiple-Mutation Theory of Carcinogenesis. Nature 1958, 181, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Mutation and Cancer: Statistical Study of Retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef]
- Albertson, D.G. Gene amplification in cancer. Trends Genet. 2006, 22, 447–455. [Google Scholar] [CrossRef]
- Verhaak, R.G.W.; Bafna, V.; Mischel, P.S. Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nat. Rev. Cancer 2019, 19, 283–288. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Alt, F.W.; Kellems, R.E.; Bertino, J.R.; Schimke, R.T. Selective multiplication of dihydrofolate reductase genes in methotrexate-resistant variants of cultured murine cells. J. Biol. Chem. 1978, 253, 1357–1370. [Google Scholar] [CrossRef]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar] [CrossRef]
- Garsed, D.W.; Marshall, O.J.; Corbin, V.D.; Hsu, A.; Di Stefano, L.; Schröder, J.; Li, J.; Feng, Z.P.; Kim, B.W.; Kowarsky, M.; et al. The Architecture and Evolution of Cancer Neochromosomes. Cancer Cell 2014, 26, 653–667. [Google Scholar] [CrossRef] [PubMed]
- Glodzik, D.; Morganella, S.; Davies, H.; Simpson, P.T.; Li, Y.; Zou, X.; Diez-Perez, J.; Staaf, J.; Alexandrov, L.B.; Smid, M.; et al. A somatic-mutational process recurrently duplicates germline susceptibility loci and tissue-specific super-enhancers in breast cancers. Nature Genet. 2017, 49, 341–348. [Google Scholar] [CrossRef]
- Menghi, F.; Barthel, F.P.; Yadav, V.; Tang, M.; Ji, B.; Tang, Z.; Carter, G.W.; Ruan, Y.; Scully, R.; Verhaak, R.G.; et al. The Tandem Duplicator Phenotype Is a Prevalent Genome-Wide Cancer Configuration Driven by Distinct Gene Mutations. Cancer Cell 2018, 34, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Martin, B.; Alvarez, E.G.; Baez-Ortega, A.; Zamora, J.; Supek, F.; Demeulemeester, J.; Santamarina, M.; Ju, Y.S.; Temes, J.; Garcia-Souto, D.; et al. Pan-cancer analysis of whole genomes identifies driver rearrangements promoted by LINE-1 retrotransposition. Nat. Genet. 2020, 52, 306–319. [Google Scholar] [CrossRef]
- Nunberg, J.H.; Kaufman, R.J.; Schimke, R.T.; Urlaub, G.; Chasin, L.A. Amplified dihydrofolate reductase genes are localized to a homogeneously staining region of a single chromosome in a methotrexate-resistant Chinese hamster ovary cell line. Proc. Natl. Acad. Sci. USA 1978, 75, 5553–5556. [Google Scholar] [CrossRef]
- Xing, R.; Zhou, Y.; Yu, J.; Yu, Y.; Nie, Y.; Luo, W.; Yang, C.; Xiong, T.; Wu, W.K.; Li, Z.; et al. Whole-genome sequencing reveals novel tandem-duplication hotspots and a prognostic mutational signature in gastric cancer. Nat. Commun. 2019, 10, 2037. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.; Yuncken, C.; Spriggs, A. Minute chromatin bodies in malignant tumours of childhood. Lancet 1965, 286, 55–58. [Google Scholar] [CrossRef]
- Carroll, S.M.; DeRose, M.L.; Gaudray, P.; Moore, C.M.; Needham-Vandevanter, D.R.; Von Hoff, D.D.; Wahl, G.M. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Mol. Cell Biol. 1988, 8, 1525–1533. [Google Scholar] [CrossRef]
- Ruiz, J.C.; Wahl, G.M. Chromosomal destabilization during gene amplification. Mol. Cell Biol. 1990, 10, 3056–3066. [Google Scholar] [CrossRef]
- Cohen, S.; Regev, A.; Lavi, S. Small polydispersed circular DNA (spcDNA) in human cells: Association with genomic instability. Oncogene 1997, 14, 977–985. [Google Scholar] [CrossRef] [PubMed]
- De Carvalho, A.C.; Kim, H.; Poisson, L.M.; Winn, M.E.; Mueller, C.; Cherba, D.; Koeman, J.; Seth, S.; Protopopov, A.; Felicella, M.; et al. Discordant inheritance of chromosomal and extrachromosomal DNA elements contributes to dynamic disease evolution in glioblastoma. Nat. Genet. 2018, 50, 708–717. [Google Scholar] [CrossRef]
- Stanfield, S.W.; Lengyel, J.A. Small circular DNA of Drosophila melanogaster: Chromosomal homology and kinetic complexity. Proc. Natl. Acad. Sci. USA 1979, 76, 6142–6146. [Google Scholar] [CrossRef]
- Shibata, Y.; Kumar, P.; Layer, R.; Willcox, S.; Gagan, J.R.; Griffith, J.D.; Dutta, A. Extrachromosomal MicroDNAs and Chromosomal Microdeletions in Normal Tissues. Science 2012, 336, 82–86. [Google Scholar] [CrossRef]
- Shoura, M.J.; Gabdank, I.; Hansen, L.; Merker, J.; Gotlib, J.; Levene, S.D.; Fire, A.Z. Intricate and Cell Type-Specific Populations of Endogenous Circular DNA (eccDNA) in Caenorhabditis elegans and Homo sapiens. G3 Genes Genomes Genet. 2017, 7, 3295–3303. [Google Scholar] [CrossRef]
- Møller, H.D.; Mohiyuddin, M.; Prada-Luengo, I.; Sailani, M.R.; Halling, J.F.; Plomgaard, P.; Maretty, L.; Hansen, A.J.; Snyder, M.P.; Pilegaard, H.; et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 2018, 9, 1069. [Google Scholar] [CrossRef]
- Paulsen, T.; Kumar, P.; Koseoglu, M.M.; Dutta, A. Discoveries of Extrachromosomal Circles of DNA in Normal and Tumor Cells. Trends Genet. 2018, 34, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Stephens, P.J.; Greenman, C.D.; Fu, B.; Yang, F.; Bignell, G.R.; Mudie, L.J.; Pleasance, E.D.; Lau, K.W.; Beare, D.; Stebbings, L.A.; et al. Massive Genomic Rearrangement Acquired in a Single Catastrophic Event during Cancer Development. Cell 2011, 144, 27–40. [Google Scholar] [CrossRef]
- Maher, C.A.; Wilson, R.K. Chromothripsis and Human Disease: Piecing Together the Shattering Process. Cell 2012, 148, 29–32. [Google Scholar] [CrossRef]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, F.H. Gradualism, punctuated equilibrium and the Origin of Species. Nature 1983, 305, 269–272. [Google Scholar] [CrossRef]
- Gould, S.j.; Eldredge, N. Punctuated equilibrium comes of age. Nature 1993, 366, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Forment, J.V.; Kaidi, A.; Jackson, S.P. Chromothripsis and cancer: Causes and consequences of chromosome shattering. Nat. Rev. Cancer 2012, 12, 663–670. [Google Scholar] [CrossRef]
- Malhotra, A.; Lindberg, M.; Faust, G.G.; Leibowitz, M.L.; Clark, R.A.; Layer, R.M.; Quinlan, A.R.; Hall, I.M. Breakpoint profiling of 64 cancer genomes reveals numerous complex rearrangements spawned by homology-independent mechanisms. Genome Res. 2013, 23, 762–776. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Leibowitz, M.L.; Pellman, D. Chromothripsis and beyond: Rapid genome evolution from complex chromosomal rearrangements. Genes Dev. 2013, 27, 2513–2530. [Google Scholar] [CrossRef]
- Sottoriva, A.; Kang, H.; Ma, Z.; Graham, T.A.; Salomon, M.P.; Zhao, J.; Marjoram, P.; Siegmund, K.; Press, M.F.; Shibata, D.; et al. A Big Bang model of human colorectal tumor growth. Nature Genet. 2015, 47, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Heng, J.; Heng, H.H. Genome chaos: Creating new genomic information essential for cancer macroevolution. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef]
- Shapiro, J.A. What can evolutionary biology learn from cancer biology? Prog. Biophys. Mol. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.J.; Stilgenbauer, L.; Moy, A.; Liu, G.; Heng, H.H. What Is Karyotype Coding and Why Is Genomic Topology Important for Cancer and Evolution? Front. Genet. 2019, 10, 104476. [Google Scholar] [CrossRef]
- Crasta, K.; Ganem, N.J.; Dagher, R.; Lantermann, A.B.; Ivanova, E.V.; Pan, Y.; Nezi, L.; Protopopov, A.; Chowdhury, D.; Pellman, D. DNA breaks and chromosome pulverization from errors in mitosis. Nature 2012, 482, 53–58. [Google Scholar] [CrossRef]
- Zhang, C.Z.; Spektor, A.; Cornils, H.; Francis, J.M.; Jackson, E.K.; Liu, S.; Meyerson, M.; Pellman, D. Chromothripsis from DNA damage in micronuclei. Nature 2015, 522, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Cortés-Ciriano, I.; Lee, J.J.K.; Xi, R.; Jain, D.; Jung, Y.L.; Yang, L.; Gordenin, D.; Klimczak, L.J.; Zhang, C.Z.; Pellman, D.S.; et al. Comprehensive analysis of chromothripsis in 2658 human cancers using whole-genome sequencing. Nature Genet. 2020, 52, 331–341. [Google Scholar] [CrossRef]
- Zack, T.I.; Schumacher, S.E.; Carter, S.L.; Cherniack, A.D.; Saksena, G.; Tabak, B.; Lawrence, M.S.; Zhang, C.Z.; Wala, J.; Mermel, C.H.; et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013, 45, 1134–1140. [Google Scholar] [CrossRef] [PubMed]
- Bielski, C.M.; Zehir, A.; Penson, A.V.; Donoghue, M.T.; Chatila, W.; Armenia, J.; Chang, M.T.; Schram, A.M.; Jonsson, P.; Bandlamudi, C.; et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018, 50, 1189–1195. [Google Scholar] [CrossRef] [PubMed]
- Davoli, T.; de Lange, T. The Causes and Consequences of Polyploidy in Normal Development and Cancer. Annu. Rev. Cell Dev. Biol. 2011, 27, 585–610. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Bandi, M.; Nitta, M.; Ivanova, E.V.; Bronson, R.T.; Pellman, D. Cytokinesis failure generating tetraploids promotes tumorigenesis in p53-null cells. Nature 2005, 437, 1043–1047. [Google Scholar] [CrossRef] [PubMed]
- Dewhurst, S.M.; McGranahan, N.; Burrell, R.A.; Rowan, A.J.; Grönroos, E.; Endesfelder, D.; Joshi, T.; Mouradov, D.; Gibbs, P.; Ward, R.L.; et al. Tolerance of whole- genome doubling propagates chromosomal instability and accelerates cancer genome evolution. Cancer Disc. 2014, 4, 175–185. [Google Scholar] [CrossRef] [PubMed]
- López, S.; Lim, E.L.; Horswell, S.; Haase, K.; Huebner, A.; Dietzen, M.; Mourikis, T.P.; Watkins, T.B.; Rowan, A.; Dewhurst, S.M.; et al. Interplay between whole-genome doubling and the accumulation of deleterious alterations in cancer evolution. Nat. Genet. 2020, 52, 283–293. [Google Scholar] [CrossRef]
- Duijf, P.H.; Schultz, N.; Benezra, R. Cancer cells preferentially lose small chromosomes. Int. J. Cancer 2013, 132, 2316–2326. [Google Scholar] [CrossRef]
- Silk, A.D.; Zasadil, L.M.; Holland, A.J.; Vitre, B.; Cleveland, D.W.; Weaver, B.A. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc. Natl. Acad. Sci. USA 2013, 110, E4134–E4141. [Google Scholar] [CrossRef] [PubMed]
- Andreassen, P.R.; Lohez, O.D.; Lacroix, F.B.; Margolis, R.L. Tetraploid State Induces p53-dependent Arrest of Nontransformed Mammalian Cells in G1. Mol. Biol. Cell 2001, 12, 1315–1328. [Google Scholar] [CrossRef] [PubMed]
- Quinton, R.J.; DiDomizio, A.; Vittoria, M.A.; Kotýnková, K.; Ticas, C.J.; Patel, S.; Koga, Y.; Vakhshoorzadeh, J.; Hermance, N.; Kuroda, T.S.; et al. Whole-genome doubling confers unique genetic vulnerabilities on tumour cells. Nature 2021, 590, 492–497. [Google Scholar] [CrossRef]
- Kim, T.M.; Xi, R.; Luquette, L.J.; Park, R.W.; Johnson, M.D.; Park, P.J. Functional genomic analysis of chromosomal aberrations in a compendium of 8000 cancer genomes. Genome Res. 2013, 23, 217–227. [Google Scholar] [CrossRef]
- Cai, H.; Kumar, N.; Bagheri, H.C.; von Mering, C.; Robinson, M.D.; Baudis, M. Chromothripsis-like patterns are recurring but heterogeneously distributed features in a survey of 22,347 cancer genome screens. BMC Genom. 2014, 15, 82. [Google Scholar] [CrossRef] [PubMed]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L.; Moorhead, P. The serial cultivation of human diploid cell strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Zhang, X.; Mar, V.; Zhou, W.; Harrington, L.; Robinson, M.O. Telomere shortening and apoptosis in telomerase-inhibited human tumor cells. Genes Dev. 1999, 13, 2388–2399. [Google Scholar] [CrossRef]
- Poole, J.C.; Andrews, L.G.; Tollefsbol, T.O. Activity, function, and gene regulation of the catalytic subunit of telomerase (hTERT). Gene 2001, 269, 1–12. [Google Scholar] [CrossRef]
- Cao, Y.; Bryan, T.M.; Reddel, R.R. Increased copy number of the TERT and TERC telomerase subunit genes in cancer cells. Cancer Sci. 2008, 99, 1092–1099. [Google Scholar] [CrossRef]
- Gay-Bellile, M.; Véronèse, L.; Combes, P.; Eymard-Pierre, E.; Kwiatkowski, F.; Dauplat, M.M.; Cayre, A.; Privat, M.; Abrial, C.; Bignon, Y.J.; et al. TERT promoter status and gene copy number gains: Effect on TERT expression and association with prognosis in breast cancer. Oncotarget 2017, 8, 77540–77551. [Google Scholar] [CrossRef]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef]
- Haber, J.E.; Debatisse, M. Gene Amplification: Yeast Takes a Turn. Cell 2006, 125, 1237–1240. [Google Scholar] [CrossRef][Green Version]
- Lee, J.J.K.; Park, S.; Park, H.; Kim, S.; Lee, J.; Lee, J.; Youk, J.; Yi, K.; An, Y.; Park, I.K.; et al. Tracing Oncogene Rearrangements in the Mutational History of Lung Adenocarcinoma. Cell 2019, 177, 1842–1857. [Google Scholar] [CrossRef]
- Tanaka, H.; Watanabe, T. Mechanisms Underlying Recurrent Genomic Amplification in Human Cancers. Trends Cancer 2020, 6, 462–477. [Google Scholar] [CrossRef]
- Martincorena, I.; Raine, K.M.; Gerstung, M.; Dawson, K.J.; Haase, K.; Van Loo, P.; Davies, H.; Stratton, M.R.; Campbell, P.J. Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 2017, 171, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Priestley, P.; Baber, J.; Lolkema, M.P.; Steeghs, N.; de Bruijn, E.; Shale, C.; Duyvesteyn, K.; Haidari, S.; van Hoeck, A.; Onstenk, W.; et al. Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 2019, 575, 210–216. [Google Scholar] [CrossRef]
- Aylon, Y.; Oren, M. p53: Guardian of ploidy. Mol. Oncol. 2011, 5, 315–323. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 54–283. [Google Scholar] [CrossRef]
- Santaguida, S.; Richardson, A.; Iyer, D.R.; M’Saad, O.; Zasadil, L.; Knouse, K.A.; Wong, Y.L.; Rhind, N.; Desai, A.; Amon, A. Chromosome Mis-segregation Generates Cell-Cycle-Arrested Cells with Complex Karyotypes that Are Eliminated by the Immune System. Dev. Cell 2017, 41, 638–651. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Carroll, P.; Martin, C.A.; Murina, O.; Fluteau, A.; Simpson, D.J.; Olova, N.; Sutcliffe, H.; Rainger, J.K.; Leitch, A.; et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 2017, 548, 461–465. [Google Scholar] [CrossRef]
- Davoli, T.; Uno, H.; Wooten, E.C.; Elledge, S.J. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017, 355, eaaf8399. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.M.; Shih, J.; Ha, G.; Gao, G.F.; Zhang, X.; Berger, A.C.; Schumacher, S.E.; Wang, C.; Hu, H.; Liu, J.; et al. Genomic and Functional Approaches to Understanding Cancer Aneuploidy. Cancer Cell 2018, 33, 676–689. [Google Scholar] [CrossRef]
- Ben-David, U.; Amon, A. Context is everything: Aneuploidy in cancer. Nat. Rev. Genet 2020, 21, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Song, C.; Tseng, G.; Ren, B.G.; LaFramboise, W.; Michalopoulos, G.; Nelson, J.; Luo, J.H. Genome Abnormalities Precede Prostate Cancer and Predict Clinical Relapse. Am. J. Pathol. 2012, 180, 2240–2248. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.P.; Liu, P.; Nelson, J.; Hamilton, R.L.; Bhargava, R.; Michalopoulos, G.; Chen, Q.; Zhang, J.; Ma, D.; Pennathur, A.; et al. Identification of recurrent fusion genes across multiple cancer types. Sci. Rep. 2019, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Nowell, P.; Hungerford, D. A minute chromosome in human chronic granulocytic leukemia. Science 1960, 1497, 147. [Google Scholar]
- Rowley, J. A New Consistent Chromosomal Abnormality in Chronic Myelogenous Leukaemia identified by Quinacrine Fluorescence and Giemsa Staining. Nature 1973, 243, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Ar-Rushdi, A.; Nishikura, K.; Erikson, J.; Watt, R.; Rovera, G.; Croce, C. Differential expression of the translocated and the untranslocated c-myc oncogene in Burkitt lymphoma. Science 1983, 222, 390–393. [Google Scholar] [CrossRef]
- Battey, J.; Moulding, C.; Taub, R.; Murphy, W.; Stewart, T.; Potter, H.; Lenoir, G.; Leder, P. The human c-myc oncogene: Structural consequences of translocation into the igh locus in Burkitt lymphoma. Cell 1983, 34, 779–787. [Google Scholar] [CrossRef]
- Shtivelman, E.; Lifshitz, B.; Gale, R.P.; Canaani, E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature 1985, 315, 550–554. [Google Scholar] [CrossRef]
- Stam, K.; Heisterkamp, N.; Grosveld, G.; de Klein, A.; Verma, R.S.; Coleman, M.; Dosik, H.; Groffen, J. Evidence of a New Chimeric bcr /c- abl mRNA in Patients with Chronic Myelocytic Leukemia and the Philadelphia Chromosome. N. Engl. J. Med. 1985, 313, 1429–1433. [Google Scholar] [CrossRef]
- Carè, A.; Cianetti, L.; Giampaolo, A.; Sposi, N.M.; Zappavigna, V.; Mavilio, F.; Alimena, G.; Amadori, S.; Mandelli, F.; Peschle, C. Translocation of c-myc into the immunoglobulin heavy-chain locus in human acute B-cell leukemia. A molecular analysis. EMBO J 1986, 5, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Rabbitts, T.H. Chromosomal translocations in human cancer. Nature 1994, 372, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Prywes, R.; Foulkes, J.G.; Baltimore, D. The minimum transforming region of v-abl is the segment encoding protein-tyrosine kinase. J. Virol. 1985, 54, 114–122. [Google Scholar] [CrossRef]
- Latysheva, N.S.; Babu, M.M. Discovering and understanding oncogenic gene fusions through data intensive computational approaches. Nucleic Acids Res. 2016, 44, 4487–4503. [Google Scholar] [CrossRef]
- Dehghannasiri, R.; Freeman, D.E.; Jordanski, M.; Hsieh, G.L.; Damljanovic, A.; Lehnert, E.; Salzman, J. Improved detection of gene fusions by applying statistical methods reveals oncogenic RNA cancer drivers. Proc. Natl. Acad. Sci. USA 2019, 116, 15524–15533. [Google Scholar] [CrossRef]
- Heydt, C.; Wölwer, C.B.; Velazquez Camacho, O.; Wagener-Ryczek, S.; Pappesch, R.; Siemanowski, J.; Rehker, J.; Haller, F.; Agaimy, A.; Worm, K.; et al. Detection of gene fusions using targeted next-generation sequencing: A comparative evaluation. BMC Med. Genomics 2021, 14, 62. [Google Scholar] [CrossRef]
- Mitelman, F.; Johansson, B.; Mertens, F. (Eds.) Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer; John Wiley & Sons: Hoboken, NJ, USA, 2021. [Google Scholar]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S.; et al. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646–1651. [Google Scholar] [CrossRef] [PubMed]
- Klijn, C.; Durinck, S.; Stawiski, E.W.; Haverty, P.M.; Jiang, Z.; Liu, H.; Degenhardt, J.; Mayba, O.; Gnad, F.; Liu, J.; et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat. Biotechnol. 2015, 33, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Ciampi, R.; Knauf, J.A.; Kerler, R.; Gandhi, M.; Zhu, Z.; Nikiforova, M.N.; Rabes, H.M.; Fagin, J.A.; Nikiforov, Y.E. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J. Clin. Investig. 2005, 115, 94–101. [Google Scholar] [CrossRef]
- Talpaz, M.; Shah, N.P.; Kantarjian, H.; Donato, N.; Nicoll, J.; Paquette, R.; Cortes, J.; O’Brien, S.; Nicaise, C.; Bleickardt, E.; et al. Dasatinib in Imatinib-Resistant Philadelphia Chromosome–Positive Leukemias. N. Engl. J. Med. 2006, 354, 2531–2541. [Google Scholar] [CrossRef] [PubMed]
- Romana, S.P.; Poirel, H.; Leconiat, M.; Flexor, M.A.; Mauchauffé, M.; Jonveaux, P.; Macintyre, E.A.; Berger, R.; Bernard, O.A. High frequency of t(12;21) in childhood B-lineage acute lymphoblastic leukemia. Blood 1995, 86, 4263–4269. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wu, N.; Liu, D.; Jin, Y. Recurrent Fusion Genes in Leukemia: An Attractive Target for Diagnosis and Treatment. Curr. Genom. 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Morris, S.W.; Valentine, V.; L, M.; Herbrick, J.A.; Cui, X.; Bouman, D.; Li, Y.; Mehta, P.K.; Nizetic, D.; et al. Fusion of two novel genes, RBM15 and MKL1, in the t(1;22)(p13;q13) of acute megakaryoblastic leukemia. Nature Genet. 2001, 28, 220–221. [Google Scholar] [CrossRef]
- Miyoshi, H.; Kozu, T.; Shimizu, K.; Enomoto, K.; Maseki, N.; Kaneko, Y.; Kamada, N.; Ohki, M. The t(8;21) translocation in acute myeloid leukemia results in production of an AML1-MTG8 fusion transcript. EMBO J. 1993, 12, 2715–2721. [Google Scholar] [CrossRef]
- De Thé, H.; Chomienne, C.; Lanotte, M.; Degos, L.; Dejean, A. The t(15;17) translocation of acute promyelocytic leukaemia fuses the retinoic acid receptor α gene to a novel transcribed locus. Nature 1990, 347, 558–561. [Google Scholar] [CrossRef]
- Liu, P.; Tarle, S.; Hajra, A.; Claxton, D.; Marlton, P.; Freedman, M.; Siciliano, M.; Collins, F. Fusion between transcription factor CBF beta/PEBP2 beta and a myosin heavy chain in acute myeloid leukemia. Science 1993, 261, 1041–1044. [Google Scholar] [CrossRef]
- Soupir, C.P.; Vergilio, J.A.; Cin, P.D.; Muzikansky, A.; Kantarjian, H.; Jones, D.; Hasserjian, R.P. Philadelphia Chromosome–Positive Acute Myeloid Leukemia. Am. J. CLin. Pathol. 2007, 127, 642–650. [Google Scholar] [CrossRef]
- Morris, S.; Kirstein, M.; Valentine, M.; Dittmer, K.; Shapiro, D.; Saltman, D.; Look, A. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science 1994, 263, 1281–1284. [Google Scholar] [CrossRef]
- Persson, M.; Andren, Y.; Mark, J.; Horlings, H.M.; Persson, F.; Stenman, G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc. Natl. Acad. Sci. USA 2009, 106, 18740–18744. [Google Scholar] [CrossRef]
- Lens, D.; Coignet, L.; Brito-Babapulle, V.; Lima, C.; Matutes, E.; Dyer, M.; Catovsky, D. B cell prolymphocytic leukaemia (B-PLL) with complex karyotype and concurrent abnormalities of the p53 and c-MYC gene. Leukemia 1999, 13, 873–876. [Google Scholar] [CrossRef]
- Einerson, R.R.; Law, M.E.; Blair, H.E.; Kurtin, P.J.; McClure, R.F.; Ketterling, R.P.; Flynn, H.C.; Dogan, A.; Remstein, E.D. Novel FISH probes designed to detect IGK-MYC and IGL-MYC rearrangements in B-cell lineage malignancy identify a new breakpoint cluster region designated BVR2. Leukemia 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Seshagiri, S.; Stawiski, E.W.; Durinck, S.; Modrusan, Z.; Storm, E.E.; Conboy, C.B.; Chaudhuri, S.; Guan, Y.; Janakiraman, V.; Jaiswal, B.S.; et al. Recurrent R-spondin fusions in colon cancer. Nature 2012, 488, 660–664. [Google Scholar] [CrossRef] [PubMed]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Tsujimoto, Y.; Finger, L.; Yunis, J.; Nowell, P.; Croce, C. Cloning of the chromosome breakpoint of neoplastic B cells with the t(14;18) chromosome translocation. Science 1984, 226, 1097–1099. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef]
- Chen, Z.H.; Yu, Y.P.; Tao, J.; Liu, S.; Tseng, G.; Nalesnik, M.; Hamilton, R.; Bhargava, R.; Nelson, J.B.; Pennathur, A.; et al. MAN2A1–FER Fusion Gene Is Expressed by Human Liver and Other Tumor Types and Has Oncogenic Activity in Mice. Gastroenterology 2017, 153, 1120–1132. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.; Chan, J.M.; Zoppoli, P.; Niola, F.; Sullivan, R.; Castano, A.; Liu, E.M.; Reichel, J.; Porrati, P.; Pellegatta, S.; et al. Transforming Fusions of FGFR and TACC Genes in Human Glioblastoma. Science 2012, 337, 1231–1235. [Google Scholar] [CrossRef]
- Davies, K.D.; Le, A.T.; Theodoro, M.F.; Skokan, M.C.; Aisner, D.L.; Berge, E.M.; Terracciano, L.M.; Cappuzzo, F.; Incarbone, M.; Roncalli, M.; et al. Identifying and Targeting ROS1 Gene Fusions in Non–Small Cell Lung Cancer. Clin. Cancer. Res. 2012, 18, 4570–4579. [Google Scholar] [CrossRef]
- Guarnerio, J.; Bezzi, M.; Jeong, J.C.; Paffenholz, S.V.; Berry, K.; Naldini, M.M.; Lo-Coco, F.; Tay, Y.; Beck, A.H.; Pandolfi, P.P. Oncogenic Role of Fusion-circRNAs Derived from Cancer-Associated Chromosomal Translocations. Cell 2016, 165, 289–302. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.i.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Salzman, J.; Marinelli, R.J.; Wang, P.L.; Green, A.E.; Nielsen, J.S.; Nelson, B.H.; Drescher, C.W.; Brown, P.O. ESRRA-C11orf20 Is a Recurrent Gene Fusion in Serous Ovarian Carcinoma. PLoS Biol. 2011, 9, e1001156. [Google Scholar] [CrossRef]
- Jones, D.T.; Kocialkowski, S.; Liu, L.; Pearson, D.M.; Bäcklund, L.M.; Ichimura, K.; Collins, V.P. Tandem Duplication Producing a Novel Oncogenic BRAF Fusion Gene Defines the Majority of Pilocytic Astrocytomas. Cancer Res. 2008, 68, 8673–8677. [Google Scholar] [CrossRef]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Mehra, R.; Rhodes, D.R.; Smith, L.R.; Roulston, D.; Helgeson, B.E.; Cao, X.; Wei, J.T.; Rubin, M.A.; Shah, R.B.; et al. TMPRSS2:ETV4 Gene Fusions Define a Third Molecular Subtype of Prostate Cancer. Cancer Res. 2006, 66, 3396–3400. [Google Scholar] [CrossRef]
- Yu, Y.P.; Ding, Y.; Chen, Z.; Liu, S.; Michalopoulos, A.; Chen, R.; Gulzar, Z.G.; Yang, B.; Cieply, K.M.; Luvison, A.; et al. Novel Fusion Transcripts Associate with Progressive Prostate Cancer. Am. J. Pathol. 2014, 184, 2840–2849. [Google Scholar] [CrossRef]
- Kroll, T.G.; Sarraf, P.; Pecciarini, L.; Chen, C.J.; Mueller, E.; Spiegelman, B.M.; Fletcher, J.A. PAX8-PPARgamma1 fusion oncogene in human thyroid carcinoma [corrected]. Science 2000, 289, 1357–1360. [Google Scholar] [CrossRef]
- Li, Y.; Chien, J.; Smith, D.I.; Ma, J. FusionHunter: Identifying fusion transcripts in cancer using paired-end RNA-seq. Bioinformatics 2011, 27, 1708–1710. [Google Scholar] [CrossRef]
- McPherson, A.; Hormozdiari, F.; Zayed, A.; Giuliany, R.; Ha, G.; Sun, M.G.F.; Griffith, M.; Heravi Moussavi, A.; Senz, J.; Melnyk, N.; et al. deFuse: An Algorithm for Gene Fusion Discovery in Tumor RNA-Seq Data. PLoS Comput. Biol. 2011, 7, e1001138. [Google Scholar] [CrossRef]
- Jia, W.; Qiu, K.; He, M.; Song, P.; Zhou, Q.; Zhou, F.; Yu, Y.; Zhu, D.; Nickerson, M.L.; Wan, S.; et al. SOAPfuse: An algorithm for identifying fusion transcripts from paired-end RNA-Seq data. Genome Biol. 2013, 14, R12. [Google Scholar] [CrossRef]
- Nicorici, D.; Şatalan, M.; Edgren, H.; Kangaspeska, S.; Murumägi, A.; Kallioniemi, O.; Virtanen, S.; Kilkku, O. FusionCatcher—A tool for finding somatic fusion genes in paired-end RNA-sequencing data. bioRxiv 2014. [Google Scholar] [CrossRef]
- Torres-García, W.; Zheng, S.; Sivachenko, A.; Vegesna, R.; Wang, Q.; Yao, R.; Berger, M.F.; Weinstein, J.N.; Getz, G.; Verhaak, R.G. PRADA: Pipeline for RNA sequencing data analysis. Bioinformatics 2014, 30, 2224–2226. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.M.; Majewski, I.J.; Oshlack, A. JAFFA: High sensitivity transcriptome-focused fusion gene detection. Genome Med. 2015, 7, 43. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Tsai, W.H.; Ding, Y.; Chen, R.; Fang, Z.; Huo, Z.; Kim, S.; Ma, T.; Chang, T.Y.; Priedigkeit, N.M.; et al. Comprehensive evaluation of fusion transcript detection algorithms and a meta-caller to combine top performing methods in paired-end RNA-seq data. Nucleic Acids Res. 2016, 44, e47. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Stransky, N.; Li, B.; Yang, X.; Tickle, T.; Bankapur, A.; Ganote, C.; Doak, T.G.; Pochet, N.; et al. STAR-Fusion: Fast and Accurate Fusion Transcript Detection from RNA-Seq. bioRxiv 2017. [Google Scholar] [CrossRef]
- Uhrig, S.; Ellermann, J.; Walther, T.; Burkhardt, P.; Fröhlich, M.; Hutter, B.; Toprak, U.H.; Neumann, O.; Stenzinger, A.; Scholl, C.; et al. Accurate and efficient detection of gene fusions from RNA sequencing data. Gen. Res. 2021, 31, 448–460. [Google Scholar] [CrossRef]
- Haas, B.J.; Dobin, A.; Li, B.; Stransky, N.; Pochet, N.; Regev, A. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol. 2019, 20, 213. [Google Scholar] [CrossRef]
- Carrara, M.; Beccuti, M.; Cavallo, F.; Donatelli, S.; Lazzarato, F.; Cordero, F.; Calogero, R.A. State of art fusion-finder algorithms are suitable to detect transcription-induced chimeras in normal tissues? BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Vo, A.D.; Qin, F.; Li, H. Comparative assessment of methods for the fusion transcripts detection from RNA-Seq data. Sci. Rep. 2016, 6, 21597. [Google Scholar] [CrossRef] [PubMed]
- Kastenhuber, E.R.; Lalazar, G.; Houlihan, S.L.; Tschaharganeh, D.F.; Baslan, T.; Chen, C.C.; Requena, D.; Tian, S.; Bosbach, B.; Wilkinson, J.E.; et al. DNAJB1–PRKACA fusion kinase interacts with β-catenin and the liver regenerative response to drive fibrolamellar hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2017, 114, 13076–13084. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, S.; Ko, S.; In, Y.h.; Moon, H.G.; Ahn, S.K.; Kim, M.K.; Lee, M.; Hwang, J.H.; Ju, Y.S.; et al. Recurrent fusion transcripts detected by whole-transcriptome sequencing of 120 primary breast cancer samples. Genes Chromosom. Cancer 2015, 54, 681–691. [Google Scholar] [CrossRef]
- Lei, J.T.; Shao, J.; Zhang, J.; Iglesia, M.; Chan, D.W.; Cao, J.; Anurag, M.; Singh, P.; He, X.; Kosaka, Y.; et al. Functional Annotation of ESR1 Gene Fusions in Estrogen Receptor-Positive Breast Cancer. Cell Rep. 2018, 24, 1434–1444. [Google Scholar] [CrossRef]
- Matissek, K.J.; Onozato, M.L.; Sun, S.; Zheng, Z.; Schultz, A.; Lee, J.; Patel, K.; Jerevall, P.L.; Saladi, S.V.; Macleay, A.; et al. Expressed Gene Fusions as Frequent Drivers of Poor Outcomes in Hormone Receptor–Positive Breast Cancer. Cancer. Disc. 2018, 8, 336–353. [Google Scholar] [CrossRef]
- Enuka, Y.; Lauriola, M.; Feldman, M.E.; Sas-Chen, A.; Ulitsky, I.; Yarden, Y. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res 2016, 44, 1370–1383. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Hansen, T.B.; Venø, M.T.; Kjems, J. Circular RNAs in cancer: Opportunities and challenges in the field. Oncogene 2018, 37, 555–565. [Google Scholar] [CrossRef]
- Su, M.; Xiao, Y.; Ma, J.; Tang, Y.; Tian, B.; Zhang, Y.; Li, X.; Wu, Z.; Yang, D.; Zhou, Y.; et al. Circular RNAs in Cancer: Emerging functions in hallmarks, stemness, resistance and roles as potential biomarkers. Mol. Cancer 2019, 18, 90. [Google Scholar] [CrossRef]
- Kumar-Sinha, C.; Tomlins, S.A.; Chinnaiyan, A.M. Recurrent gene fusions in prostate cancer. Nat. Rev. Cancer 2008, 8, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Prensner, J.R.; Chinnaiyan, A.M. Oncogenic gene fusions in epithelial carcinomas. Curr. Opin. Genet. Dev. 2009, 19, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Edwards, P.A. Fusion genes and chromosome translocations in the common epithelial cancers. J. Pathol. 2010, 220, 244–254. [Google Scholar] [CrossRef]
- Yu, J.; Yu, J.; Mani, R.S.; Cao, Q.; Brenner, C.J.; Cao, X.; Wang, X.; Wu, L.; Li, J.; Hu, M.; et al. An Integrated Network of Androgen Receptor, Polycomb, and TMPRSS2-ERG Gene Fusions in Prostate Cancer Progression. Cancer Cell 2010, 17, 443–454. [Google Scholar] [CrossRef]
- Demichelis, F.; Fall, K.; Perner, S.; Andrén, O.; Schmidt, F.; Setlur, S.R.; Hoshida, Y.; Mosquera, J.M.; Pawitan, Y.; Lee, C.; et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene 2007, 26, 4596–4599. [Google Scholar] [CrossRef]
- Nam, R.K.; Sugar, L.; Wang, Z.; Yang, W.; Kitching, R.; Klotz, L.H.; Venkateswaran, V.; Narod, S.A.; Seth, A. Expression of TMPRSS2:ERG gene fusion in prostate cancer cells is an important prognostic factor for cancer progression. Cancer Biol. Ther. 2007, 6, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Attard, G.; Clark, J.; Ambroisine, L.; Fisher, G.; Kovacs, G.; Flohr, P.; Berney, D.; Foster, C.S.; Fletcher, A.; Gerald, W.L.; et al. Duplication of the fusion of TMPRSS2 to ERG sequences identifies fatal human prostate cancer. Oncogene 2008, 27, 253–263. [Google Scholar] [CrossRef]
- John, J.S.; Powell, K.; LaComb, M.K.C. TMPRSS2-ERG Fusion Gene Expression in Prostate Tumor Cells and Its Clinical and Biological Significance in Prostate Cancer Progression. J. Cancer. Sci. Ther. 2012, 04. [Google Scholar] [CrossRef]
- Hägglöf, C.; Hammarsten, P.; Strömvall, K.; Egevad, L.; Josefsson, A.; Stattin, P.; Granfors, T.; Bergh, A. TMPRSS2-ERG Expression Predicts Prostate Cancer Survival and Associates with Stromal Biomarkers. PLoS ONE 2014, 9, e86824. [Google Scholar] [CrossRef]
- Song, C.; Chen, H. Predictive significance of TMRPSS2-ERG fusion in prostate cancer: A meta-analysis. Cancer Cell Int. 2018, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Hu, Y.; Loo, S.K.; Tan, Y.; Bhargava, R.; Lewis, M.T.; Wang, X.S. Landscape analysis of adjacent gene rearrangements reveals BCL2L14–ETV6 gene fusions in more aggressive triple-negative breast cancer. Proc. Natl. Acad. Sci. USA 2020, 117, 9912–9921. [Google Scholar] [CrossRef]
- Bao, Z.S.; Chen, H.M.; Yang, M.Y.; Zhang, C.B.; Yu, K.; Ye, W.L.; Hu, B.Q.; Yan, W.; Zhang, W.; Akers, J.; et al. RNA-seq of 272 gliomas revealed a novel, recurrent PTPRZ1-MET fusion transcript in secondary glioblastomas. Gen. Res. 2014, 24, 1765–1773. [Google Scholar] [CrossRef]
- Hao, Q.L.; Heisterkamp, N.; Groffen, J. Isolation and sequence analysis of a novel human tyrosine kinase gene. Mol. Cell Biol. 1989, 9, 1587–1593. [Google Scholar] [CrossRef]
- De Braekeleer, E.; Douet-Guilbert, N.; Rowe, D.; Bown, N.; Morel, F.; Berthou, C.; Férec, C.; De Braekeleer, M. ABL1 fusion genes in hematological malignancies: A review. Eur. J. Haematol. 2011, 86, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Parker, B.C.; Engels, M.; Annala, M.; Zhang, W. Emergence of FGFR family gene fusions as therapeutic targets in a wide spectrum of solid tumours. J. Pathol. 2014, 232, 4–15. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Chmielecki, J.; Gay, L.; Johnson, A.; Chudnovsky, J.; Yelensky, R.; Lipson, D.; Ali, S.M.; Elvin, J.A.; et al. The distribution of BRAF gene fusions in solid tumors and response to targeted therapy. Int. J. Cancer 2016, 138, 881–890. [Google Scholar] [CrossRef]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Esposito Abate, R.; Rachiglio, A.M.; Maiello, M.R.; Esposito, C.; Schettino, C.; Izzo, F.; Nasti, G.; Normanno, N. FGFR Fusions in Cancer: From Diagnostic Approaches to Therapeutic Intervention. Int. J. Mol. Sci. 2020, 21, 6856. [Google Scholar] [CrossRef]
- Chmielecki, J.; Peifer, M.; Jia, P.; Socci, N.D.; Hutchinson, K.; Viale, A.; Zhao, Z.; Thomas, R.K.; Pao, W. Targeted next-generation sequencing of DNA regions proximal to a conserved GXGXXG signaling motif enables systematic discovery of tyrosine kinase fusions in cancer. Nucleic. Acids. Res. 2010, 38, 6985–6996. [Google Scholar] [CrossRef]
- Stransky, N.; Cerami, E.; Schalm, S.; Kim, J.L.; Lengauer, C. The landscape of kinase fusions in cancer. Nat. Commun. 2014, 5, 4846. [Google Scholar] [CrossRef] [PubMed]
- Schram, A.M.; Chang, M.T.; Jonsson, P.; Drilon, A. Fusions in solid tumours: Diagnostic strategies, targeted therapy, and acquired resistance. Nat. Rev. Clin. Oncol. 2017, 14, 735–748. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.M.; Su, F.; Kalyana-Sundaram, S.; Khazanov, N.; Ateeq, B.; Cao, X.; Lonigro, R.J.; Vats, P.; Wang, R.; Lin, S.F.; et al. Identification of Targetable FGFR Gene Fusions in Diverse Cancers. Cancer Disc. 2013, 3, 636–647. [Google Scholar] [CrossRef] [PubMed]
- McDermott, U.; Iafrate, A.J.; Gray, N.S.; Shioda, T.; Classon, M.; Maheswaran, S.; Zhou, W.; Choi, H.G.; Smith, S.L.; Dowell, L.; et al. Genomic Alterations of Anaplastic Lymphoma Kinase May Sensitize Tumors to Anaplastic Lymphoma Kinase Inhibitors. Cancer Res. 2008, 68, 3389–3395. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, K.; Soda, M.; Togashi, Y.; Suzuki, R.; Sakata, S.; Hatano, S.; Asaka, R.; Hamanaka, W.; Ninomiya, H.; Uehara, H.; et al. RET, ROS1 and ALK fusions in lung cancer. Nat. Med. 2012, 18, 378–381. [Google Scholar] [CrossRef]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and Safety of a Specific Inhibitor of the BCR-ABL Tyrosine Kinase in Chronic Myeloid Leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Galkin, A.V.; Melnick, J.S.; Kim, S.; Hood, T.L.; Li, N.; Li, L.; Xia, G.; Steensma, R.; Chopiuk, G.; Jiang, J.; et al. Identification of NVP-TAE684, a potent, selective, and efficacious inhibitor of NPM-ALK. Proc. Natl. Acad. Sci. USA 2007, 104, 270–275. [Google Scholar] [CrossRef]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of Larotrectinib in TRK Fusion–Positive Cancers in Adults and Children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Meador, C.B.; Hata, A.N. Acquired resistance to targeted therapies in NSCLC: Updates and evolving insights. Pharmacol. Ther. 2020, 210, 107522. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Viossat, Y.; Noble, R. A theoretical analysis of tumour containment. Nat. Ecol. Evol. 2021, 5, 826–835. [Google Scholar] [CrossRef]
- Pemovska, T.; Johnson, E.; Kontro, M.; Repasky, G.A.; Chen, J.; Wells, P.; Cronin, C.N.; McTigue, M.; Kallioniemi, O.; Porkka, K.; et al. Axitinib effectively inhibits BCR-ABL1(T315I) with a distinct binding conformation. Nature 2015, 519, 102–105. [Google Scholar] [CrossRef]
- Nimmanapalli, R.; Bhalla, K. Novel targeted therapies for Bcr–Abl positive acute leukemias: Beyond STI571. Oncogene 2002, 21, 8584–8590. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keating, G.M. Dasatinib: A Review in Chronic Myeloid Leukaemia and Ph+ Acute Lymphoblastic Leukaemia. Drugs 2017, 77, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; Giles, F.; Gattermann, N.; Bhalla, K.; Alimena, G.; Palandri, F.; Ossenkoppele, G.J.; Nicolini, F.E.; O’Brien, S.G.; Litzow, M.; et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome–positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 2007, 110, 3540–3546. [Google Scholar] [CrossRef]
- Cui, J.J.; Tran-Dubé, M.; Shen, H.; Nambu, M.; Kung, P.P.; Pairish, M.; Jia, L.; Meng, J.; Funk, L.; Botrous, I.; et al. Structure Based Drug Design of Crizotinib (PF-02341066), a Potent and Selective Dual Inhibitor of Mesenchymal–Epithelial Transition Factor (c-MET) Kinase and Anaplastic Lymphoma Kinase (ALK). J. Med. Chem. 2011, 54, 6342–6363. [Google Scholar] [CrossRef] [PubMed]
- King, M.C.; Wilson, A.C. Evolution at two levels in humans and chimpanzees. Science 1975, 188, 107–116. [Google Scholar] [CrossRef]
- Brawand, D.; Soumillon, M.; Necsulea, A.; Julien, P.; Csardi, G.; Harrigan, P.; Weier, M.; Liechti, A.; Aximu-Petri, A.; Kircher, M.; et al. The evolution of gene expression levels in mammalian organs. Nature 2011, 478, 343–348. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Cancer Type | Fusion Gene(s) | References |
|---|---|---|
| Acute lymphoblastic leukaemia | BCR-ABL1, ETV6-RUNX1, TCF3-PBX1 | [106,107,108] |
| Acute megakaryoblastic leukaemia | RBM15–MKL1 | [109] |
| Acute myeloid leukaemia | RUNX1-RUNX1T1(AML1-MTG8), PML-RARA, CBFB-MYH11, BCR-ABL1, RBM15–MKL1 | [15,110,111,112,113] |
| Anaplastic large T-cell lymphoma | NPM1–ALK | [114] |
| Breast carcinoma | TRMT11-GRIK2, CCNH-C5orf30, ETV6–NTRK3, ODZ4–NRG1, TBL1XR1–RGS17, MYB-NFIB, MAST-fusions, NOTCH-fusions | [15,89,103,115] |
| Burkitt lymphoma | IGH–MYC, IGK–MYC, IGL–MYC | [92,116,117] |
| Chronic myeloid leukaemia | BCR-ABL1 | [94] |
| Colorectal carcinoma | TRMT11-GRIK2, CCNH-C5orf30, RSPO2-EIF3E, RSPO2-PTPRK | [89,118] |
| Ewing’s sarcoma | EWSR1-FLI1 | [119] |
| Fibrosarcoma | ETV6–NTRK3 | [15] |
| Follicular lymphoma | BCL2-IGH | [120,121] |
| Glioblastoma multiforme | TRMT11-GRIK2, CCNH-C5orf30, MAN2A1-FER, FGFR3-TACC3, FIG-ROS1 | [89,122,123,124] |
| Hepatocellular carcinoma | TRMT11-GRIK2, CCNH-C5orf30, MAN2A1-FER | [89,122] |
| Lung cancer | TRMT11-GRIK2, CCNH-C5orf30, EML4-ALK1, MAN2A1-FER, FIG-ROS1 | [89,122,124,125,126] |
| Oesophageal adenocarcinoma | TRMT11-GRIK2, CCNH-C5orf30, MAN2A1-FER | [89,122] |
| Ovarian adenocarcinoma | TRMT11-GRIK2, CCNH-C5orf30, MAN2A1-FER, ESRRA-C11orf20, FIG-ROS1 | [89,122,124,127] |
| Pilocytic astrocytoma | BRAF-KIAA1549 | [128] |
| Prostate carcinoma | TMPRSS2-ERG, TMPRSS2-ETV1, TMPRSS2-ETV4, MAN2A1-FER, TRMT11-GRIK2, SLC45A2-AMACR, TMEM135-CCDC67, MTOR-TP53BP1, CCNH-C5orf30, RPS10–HPR | [15,129,130,131] |
| Thyroid carcinoma | APAK9-BRAF, RET–CCDC6, PAX8–PPARG, TFG–NTRK1, TPM3–NTRK1 | [15,105,132] |
| Fusion Gene | Cancer Type(s) | Therapy/Drug | References |
|---|---|---|---|
| BCR-ABL1 | Acute lymphoblastic leukaemia, acute myeloid leukaemia, chronic myeloid leukaemia | imatinib, axitinib, dasatinib, nilotinib, arsenic trioxide, ponatibib | [108,176,182,183,184,185] |
| ALK-fusions | Anaplastic large T-cell lymphoma | NVP-TAE684, crizotinib | [168,177,186] |
| NRTK-fusions | Secretory breast carcinoma, mammary analogue secretory carcinoma, congenital mesoblastic nephroma, infantile fibrosarcoma, thyroid cancer, melanoma, breast cancer | larotrectinib, entrectinib, LOXO-195, TPX-0005 | [168,178] |
| ROS1-fusions | Non-small cell lung cancer | entrectinib | [124,168,177,186] |
| PML-RARA | Acute promyelocytic leukemia | All-trans retinoic acid, arsenic trioxide | [108] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Glenfield, C.; Innan, H. Gene Duplication and Gene Fusion Are Important Drivers of Tumourigenesis during Cancer Evolution. Genes 2021, 12, 1376. https://doi.org/10.3390/genes12091376
Glenfield C, Innan H. Gene Duplication and Gene Fusion Are Important Drivers of Tumourigenesis during Cancer Evolution. Genes. 2021; 12(9):1376. https://doi.org/10.3390/genes12091376
Chicago/Turabian StyleGlenfield, Cian, and Hideki Innan. 2021. "Gene Duplication and Gene Fusion Are Important Drivers of Tumourigenesis during Cancer Evolution" Genes 12, no. 9: 1376. https://doi.org/10.3390/genes12091376
APA StyleGlenfield, C., & Innan, H. (2021). Gene Duplication and Gene Fusion Are Important Drivers of Tumourigenesis during Cancer Evolution. Genes, 12(9), 1376. https://doi.org/10.3390/genes12091376