
The Role of the Rad55–Rad57 Complex in DNA Repair

{kind=link}
{kind=link}
Abstract
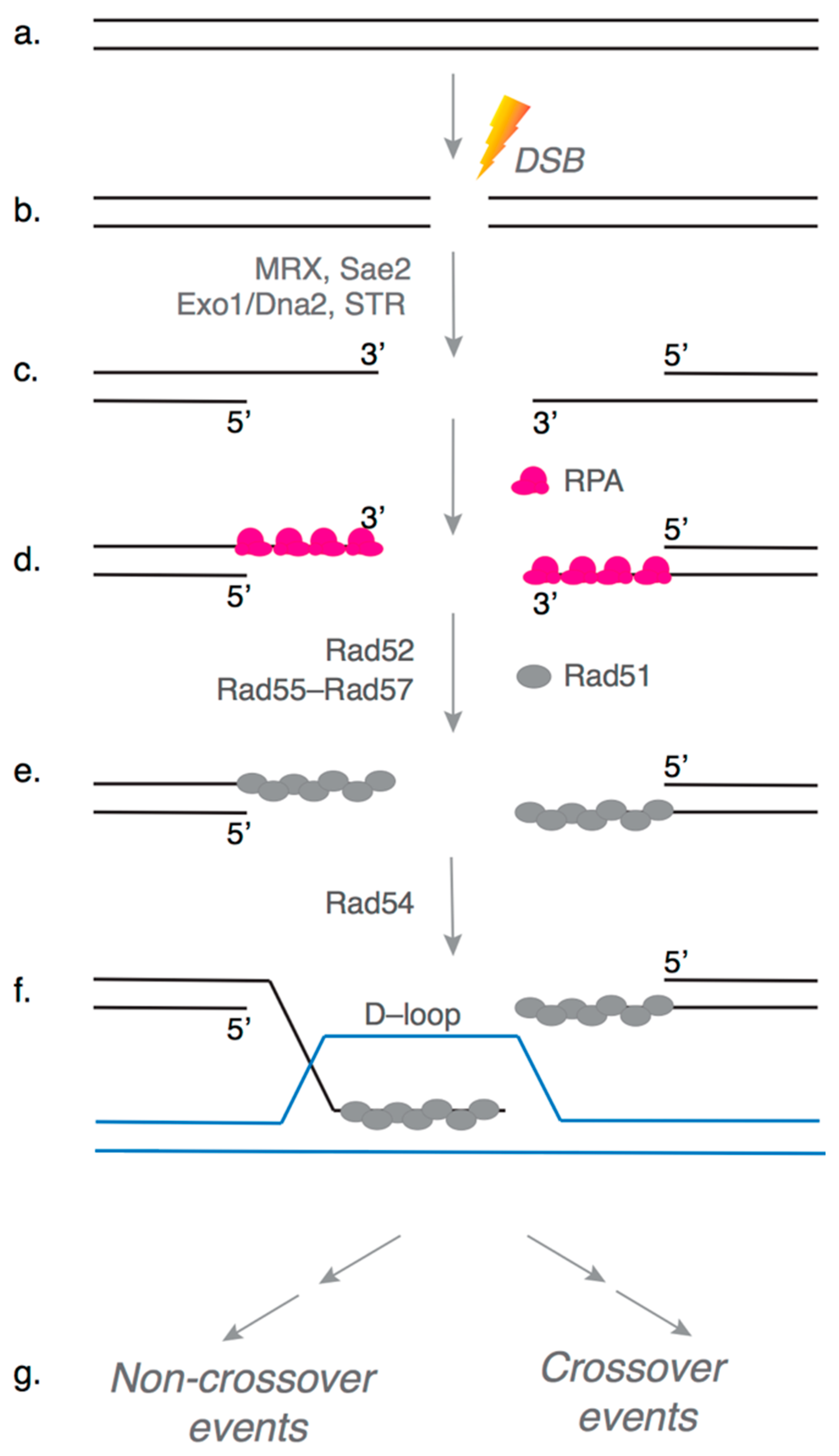
:1. Introduction
1.1. Mediator Proteins in HR
1.2. Rad51 Paralogs
2. Rad55–Rad57: Early Clues to Their Function in HR
3. Cellular Defects in rad55 rad57 Mutants
3.1. Suppression of Defects in RAD55 RAD57 Mutants
3.2. Cold-Sensitive Phenotypes in RAD55 and RAD57 Mutants
3.3. Role of Rad55–Rad57 in Spontaneous Recombination
4. Regulation of Rad55–Rad57
5. Molecular Role of Rad55–Rad57 in HR
6. Single-Molecule Perspective on Rad55–Rad57 Function
7. Open Questions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of Eukaryotic Homologous Recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [Green Version]
- Kowalczykowski, S.C. An Overview of the Molecular Mechanisms of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016410. [Google Scholar] [CrossRef] [Green Version]
- Sung, P.; Klein, H. Mechanism of Homologous Recombination: Mediators and Helicases Take on Regulatory Functions. Nat. Rev. Mol. Cell Biol. 2006, 7, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Prakash, R.; Zhang, Y.; Feng, W.; Jasin, M. Homologous Recombination and Human Health: The Roles of Brca1, Brca2, and Associated Proteins. Cold Spring Harb. Perspect. Biol. 2015, 7, a016600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, A.; Haber, J.E. Sources of DNA Double-Strand Breaks and Models of Recombinational DNA Repair. Cold Spring Harb. Perspect. Biol. 2014, 6, a016428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Symington, L.S. Mechanism and Regulation of DNA End Resection in Eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crickard, J.B.; Moevus, C.J.; Kwon, Y.; Sung, P.; Greene, E.C. Rad54 Drives Atp Hydrolysis-Dependent DNA Sequence Alignment during Homologous Recombination. Cell 2020, 181, 1380–1394. [Google Scholar] [CrossRef]
- Brush, G.S.; Morrow, D.M.; Hieter, P.; Kelly, T.J. The Atm Homologue Mec1 Is Required for Phosphorylation of Replication Protein a in Yeast. Proc. Natl. Acad. Sci. USA 1996, 93, 15075–15080. [Google Scholar] [CrossRef] [Green Version]
- Sung, P. Yeast Rad55 and Rad57 Proteins Form a Heterodimer That Functions with Replication Protein a to Promote DNA Strand Exchange by Rad51 Recombinase. Genes Dev. 1997, 11, 1111–1121. [Google Scholar] [CrossRef] [Green Version]
- Sung, P. Catalysis of ATP-Dependent Homologous DNA Pairing and Strand Exchange by Yeast Rad51 Protein. Science 1994, 265, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Sung, P.; Robberson, D.L. DNA Strand Exchange Mediated by a Rad51-Ssdna Nucleoprotein Filament with Polarity Opposite to That of Reca. Cell 1995, 82, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Gaines, W.A.; Godin, S.K.; Kabbinavar, F.F.; Rao, T.; Van Demark, A.P.; Sung, P.; Bernstein, K.A. Promotion of Presynaptic Filament Assembly by the Ensemble of S. Cerevisiae Rad51 Paralogues with Rad52. Nat. Commun. 2015, 6, 7834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Renault, L.; Veaute, X.; Fabre, F.; Stahlberg, H.; Heyer, W.D. Rad51 Paralogues Rad55-Rad57 Balance the Antirecombinase Srs2 in Rad51 Filament Formation. Nature 2011, 479, 245–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.R.; Bernstein, K.A. RAD-Ical New Insights into Rad51 Regulation. Genes 2018, 9, 629. [Google Scholar] [CrossRef] [Green Version]
- Sung, P. Function of Yeast Rad52 Protein as a Mediator between Replication Protein a and the Rad51 Recombinase. J. Biol. Chem. 1997, 272, 28194–28197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, A.; Ogawa, T. Stimulation by Rad52 of Yeast Rad51-Mediated Recombination. Nature 1998, 391, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Bonilla, B.; Hengel, S.R.; Grundy, M.K.; Bernstein, K.A. Rad51 Gene Family Structure and Function. Annu. Rev. Genet. 2020, 54, 25–46. [Google Scholar] [CrossRef]
- Lin, Z.; Kong, H.; Nei, M.; Ma, H. Origins and Evolution of the Reca/Rad51 Gene Family: Evidence for Ancient Gene Duplication and Endosymbiotic Gene Transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 10328–10333. [Google Scholar] [CrossRef] [Green Version]
- Thacker, J. A Surfeit of Rad51-Like Genes? Trends Genet. 1999, 15, 166–168. [Google Scholar] [CrossRef]
- Game, J.C.; Mortimer, R.K. A Genetic Study of X-Ray Sensitive Mutants in Yeast. Mutat. Res. 1974, 24, 281–292. [Google Scholar] [CrossRef]
- Lovett, S.T. Sequence of the Rad55 Gene of Saccharomyces Cerevisiae: Similarity of Rad55 to Prokaryotic Reca and Other Reca-Like Proteins. Gene 1994, 142, 103–106. [Google Scholar] [CrossRef]
- Kans, J.A.; Mortimer, R.K. Nucleotide Sequence of the Rad57 Gene of Saccharomyces Cerevisiae. Gene 1991, 105, 139–140. [Google Scholar] [CrossRef]
- Hays, S.L.; Firmenich, A.A.; Berg, P. Complex Formation in Yeast Double-Strand Break Repair: Participation of Rad51, Rad52, Rad55, and Rad57 Proteins. Proc. Natl. Acad. Sci. USA 1995, 92, 6925–6929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.D.; Symington, L.S. Functional Differences and Interactions among the Putative Reca Homologs Rad51, Rad55, and Rad57. Mol. Cell Biol. 1995, 15, 4843–4850. [Google Scholar] [CrossRef] [Green Version]
- Donovan, J.W.; Milne, G.T.; Weaver, D.T. Homotypic and Heterotypic Protein Associations Control Rad51 Function in Double-Strand Break Repair. Genes Dev. 1994, 8, 2552–2562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovett, S.T.; Mortimer, R.K. Characterization of Null Mutants of the Rad55 Gene of Saccharomyces Cerevisiae: Effects of Temperature, Osmotic Strength and Mating Type. Genetics 1987, 116, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Rothstein, R. Rothstein. Cell Biology of Mitotic Recombination. Cold Spring Harb. Perspect. Biol. 2015, 7, a016535. [Google Scholar] [CrossRef] [Green Version]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA Damage Response: Spatiotemporal Relationships among Checkpoint and Repair Proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, C.W.; Fortin, G.S.; Peterson, S.E.; Symington, L.S. The Rad51-K191r Atpase-Defective Mutant Is Impaired for Presynaptic Filament Formation. Mol. Cell Biol. 2006, 26, 9544–9554. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.W.; Mozlin, A.M.; Symington, L.S. Suppression of the Double-Strand-Break-Repair Defect of the Saccharomyces Cerevisiae Rad57 Mutant. Genetics 2009, 181, 1195–1206. [Google Scholar] [CrossRef] [Green Version]
- Gasior, S.L.; Wong, A.K.; Kora, Y.; Shinohara, A.; Bishop, D.K. Rad52 Associates with Rpa and Functions with Rad55 and Rad57 to Assemble Meiotic Recombination Complexes. Genes Dev. 1998, 12, 2208–2221. [Google Scholar] [CrossRef] [Green Version]
- Sugawara, N.; Wang, X.; Haber, J.E. In Vivo Roles of Rad52, Rad54, and Rad55 Proteins in Rad51-Mediated Recombination. Mol. Cell 2003, 12, 209–219. [Google Scholar] [CrossRef]
- Roy, U.; Kwon, Y.; Marie, L.; Symington, L.; Sung, P.; Lisby, M.; Greene, E.C. The Rad51 Paralog Complex Rad55-Rad57 Acts as a Molecular Chaperone During Homologous Recombination. Mol. Cell 2021, 81, 1043–1057. [Google Scholar] [CrossRef] [PubMed]
- Fortin, G.S.; Symington, L.S. Mutations in Yeast Rad51 That Partially Bypass the Requirement for Rad55 and Rad57 in DNA Repair by Increasing the Stability of Rad51-DNA Complexes. EMBO J. 2002, 21, 3160–3170. [Google Scholar] [CrossRef] [Green Version]
- Mozlin, A.M.; Fung, C.W.; Symington, L.S. Role of the Saccharomyces Cerevisiae Rad51 Paralogs in Sister Chromatid Recombination. Genetics 2008, 178, 113–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, L.; Van Komen, S.; Li, Y.; Villemain, J.; Reddy, M.S.; Klein, H.; Ellenberger, T.; Sung, P. DNA Helicase Srs2 Disrupts the Rad51 Presynaptic Filament. Nature 2003, 423, 305–309. [Google Scholar] [CrossRef]
- Veaute, X.; Jeusset, J.; Soustelle, C.; Kowalczykowski, S.C.; Le Cam, E.; Fabre, F. The Srs2 Helicase Prevents Recombination by Disrupting Rad51 Nucleoprotein Filaments. Nature 2003, 423, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Antony, E.; Tomko, E.J.; Xiao, Q.; Krejci, L.; Lohman, T.M.; Ellenberger, T. Srs2 Disassembles Rad51 Filaments by a Protein-Protein Interaction Triggering Atp Turnover and Dissociation of Rad51 from DNA. Mol. Cell 2009, 35, 105–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, Y.; Antony, E.; Doganay, S.; Koh, H.R.; Lohman, T.M.; Myong, S. Srs2 Prevents Rad51 Filament Formation by Repetitive Motion on DNA. Nat. Commun. 2013, 4, 2281. [Google Scholar] [CrossRef] [Green Version]
- Scheraga, H.A.; Nemethy, G.; Steinberg, I.Z. The Contribution of Hydrophobic Bonds to the Thermal Stability of Protein Conformations. J. Biol. Chem. 1962, 237, 2506–2508. [Google Scholar] [CrossRef]
- Kauzmann, W. Some Factors in the Interpretation of Protein Denaturation. Adv. Protein Chem. 1959, 14, 1–63. [Google Scholar] [PubMed]
- Rattray, A.J.; Shafer, B.K.; McGill, C.B.; Strathern, J.N. The Roles of Rev3 and Rad57 in Double-Strand-Break-Repair-Induced Mutagenesis of Saccharomyces Cerevisiae. Genetics 2002, 162, 1063–1077. [Google Scholar] [CrossRef]
- Weinert, T.A.; Kiser, G.L.; Hartwell, L.H. Mitotic Checkpoint Genes in Budding Yeast and the Dependence of Mitosis on DNA Replication and Repair. Genes Dev. 1994, 8, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, Y.; Desany, B.A.; Jones, W.J.; Liu, Q.; Wang, B.; Elledge, S.J. Regulation of Rad53 by the Atm-Like Kinases Mec1 and Tel1 in Yeast Cell Cycle Checkpoint Pathways. Science 1996, 271, 357–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashkirov, V.I.; King, J.S.; Bashkirova, E.V.; Schmuckli-Maurer, J.; Heyer, W.D. DNA Repair Protein Rad55 Is a Terminal Substrate of the DNA Damage Checkpoints. Mol. Cell Biol. 2000, 20, 4393–4404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janke, R.; Herzberg, K.; Rolfsmeier, M.; Mar, J.; Bashkirov, V.I.; Haghnazari, E.; Cantin, G.; Yates, J.R., 3rd; Heyer, W.D. A Truncated DNA-Damage-Signaling Response Is Activated after Dsb Formation in the G1 Phase of Saccharomyces Cerevisiae. Nucleic Acids Res. 2010, 38, 2302–2313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashkirov, V.I.; Herzberg, K.; Haghnazari, E.; Vlasenko, A.S.; Heyer, W.D. DNA Damage-Induced Phosphorylation of Rad55 Protein as a Sentinel for DNA Damage Checkpoint Activation in S. Cerevisiae. Methods Enzym. 2006, 409, 166–182. [Google Scholar]
- Janke, R.; Kong, J.; Braberg, H.; Cantin, G.; Yates, J.R., 3rd; Krogan, N.J.; Heyer, W.D. Nonsense-Mediated Decay Regulates Key Components of Homologous Recombination. Nucleic Acids Res. 2016, 44, 5218–5230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herzberg, K.; Bashkirov, V.I.; Rolfsmeier, M.; Haghnazari, E.; McDonald, W.H.; Anderson, S.; Bashkirova, E.V.; Yates, J.R., 3rd; Heyer, W.D. Phosphorylation of Rad55 on Serines 2, 8, and 14 Is Required for Efficient Homologous Recombination in the Recovery of Stalled Replication Forks. Mol. Cell Biol. 2006, 26, 8396–8409. [Google Scholar] [CrossRef] [Green Version]
- Taylor, M.R.G.; Spirek, M.; Chaurasiya, K.R.; Ward, J.D.; Carzaniga, R.; Yu, X.; Egelman, E.H.; Collinson, L.M.; Rueda, D.; Krejci, L.; et al. Rad51 Paralogs Remodel Pre-Synaptic Rad51 Filaments to Stimulate Homologous Recombination. Cell 2015, 162, 271–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin, S.; Wier, A.; Kabbinavar, F.; Bratton-Palmer, D.S.; Ghodke, H.; Van Houten, B.; Van Demark, A.P.; Bernstein, K.A. The Shu Complex Interacts with Rad51 through the Rad51 Paralogues Rad55-Rad57 to Mediate Error-Free Recombination. Nucleic Acids Res. 2013, 41, 4525–4534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.C.; Kowalczykowski, S.C. Mechanics and Single-Molecule Interrogation of DNA Recombination. Annu. Rev. Biochem. 2016, 85, 193–226. [Google Scholar] [CrossRef] [Green Version]
- Candelli, A.; Modesti, M.; Peterman, E.J.; Wuite, G.J. Single-Molecule Views on Homologous Recombination. Q. Rev. Biophys. 2013, 46, 323–348. [Google Scholar] [CrossRef] [PubMed]
- Kaniecki, K.; De Tullio, L.; Greene, E.C. A Change of View: Homologous Recombination at Single-Molecule Resolution. Nat. Rev. Genet. 2018, 19, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Greene, E.C.; Wind, S.; Fazio, T.; Gorman, J.; Visnapuu, M.L. DNA Curtains for High-Throughput Single-Molecule Optical Imaging. Methods Enzymol. 2010, 47, 2293–2315. [Google Scholar]
- Gibb, B.; Silverstein, T.D.; Finkelstein, I.J.; Greene, E.C. Single-Stranded DNA Curtains for Real-Time Single-Molecule Visualization of Protein-Nucleic Acid Interactions. Anal. Chem. 2012, 84, 7607–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.J.; Steinfeld, J.B.; Greene, E.C. Single-Stranded DNA Curtains for Studying Homologous Recombination. Methods Enzym. 2017, 582, 193–219. [Google Scholar]
- De Tullio, L.; Kaniecki, K.; Greene, E.C. Single-Stranded DNA Curtains for Studying the Srs2 Helicase Using Total Internal Reflection Fluorescence Microscopy. Methods Enzymol. 2018, 600, 407–437. [Google Scholar] [PubMed]
- Crickard, J.B.; Kaniecki, K.; Kwon, Y.; Sung, P.; Lisby, M.; Greene, E.C. Regulation of Hed1 and Rad54 Binding During Maturation of the Meiosis-Specific Presynaptic Complex. EMBO J. 2018, 37, e98728. [Google Scholar] [CrossRef]
- Belan, O.; Barroso, C.; Kaczmarczyk, A.; Anand, R.; Federico, S.; O’Reilly, N.; Newton, M.D.; Maeots, E.; Enchev, R.I.; Martinez-Perez, E.; et al. Single-molecule analysis reveals cooperative stimulation of Rad51 filament nucleation and growth by mediator proteins. Mol. Cell 2021, 81, 1058–1073. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roy, U.; Greene, E.C. The Role of the Rad55–Rad57 Complex in DNA Repair. Genes 2021, 12, 1390. https://doi.org/10.3390/genes12091390
Roy U, Greene EC. The Role of the Rad55–Rad57 Complex in DNA Repair. Genes. 2021; 12(9):1390. https://doi.org/10.3390/genes12091390
Chicago/Turabian StyleRoy, Upasana, and Eric C. Greene. 2021. "The Role of the Rad55–Rad57 Complex in DNA Repair" Genes 12, no. 9: 1390. https://doi.org/10.3390/genes12091390
APA StyleRoy, U., & Greene, E. C. (2021). The Role of the Rad55–Rad57 Complex in DNA Repair. Genes, 12(9), 1390. https://doi.org/10.3390/genes12091390