
Metagenomic Analyses of Plant Growth-Promoting and Carbon-Cycling Genes in Maize Rhizosphere Soils with Distinct Land-Use and Management Histories



Abstract
:1. Introduction
2. Materials and Methods
2.1. Soil Sampling and Sites Description
2.2. Physicochemical Analyses of Soil Samples
2.3. Metagenomic DNA Extraction and Sequencing
2.4. Metagenome Sequence Processing and Analysis of Data
3. Results
3.1. Soil Chemical Analysis
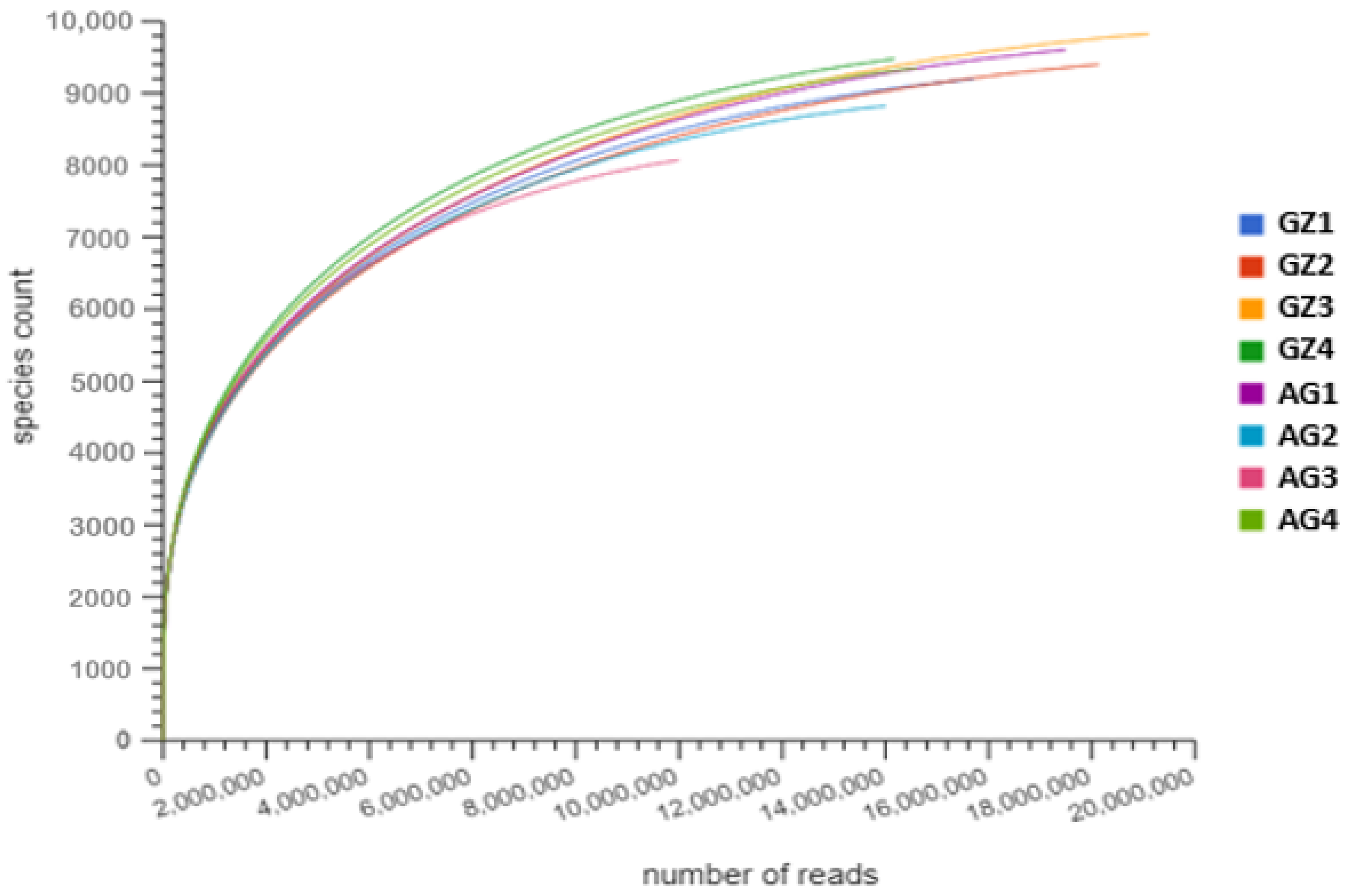
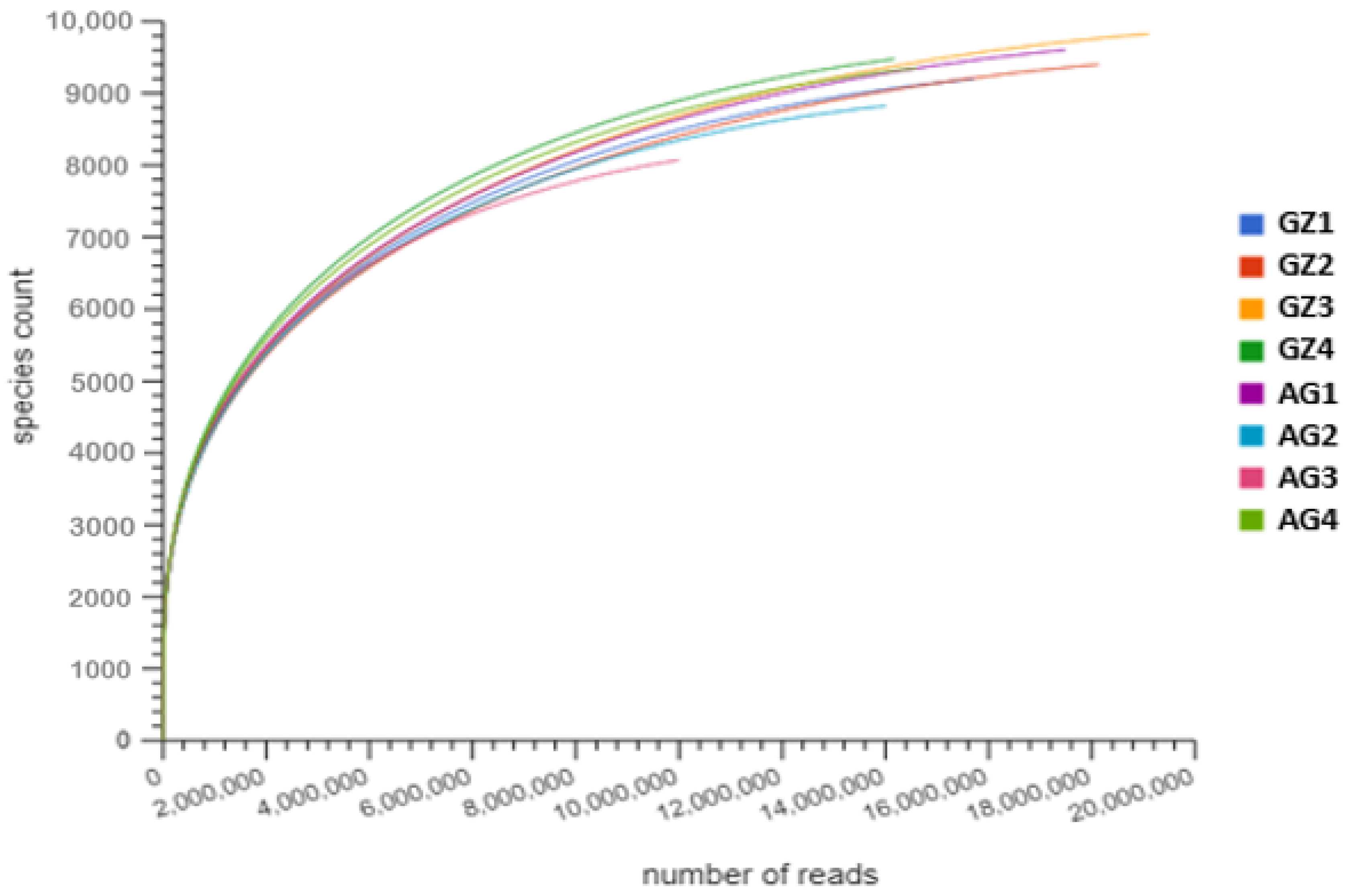
3.2. Assembly and Analysis of Shotgun Metagenomic Sequence Data
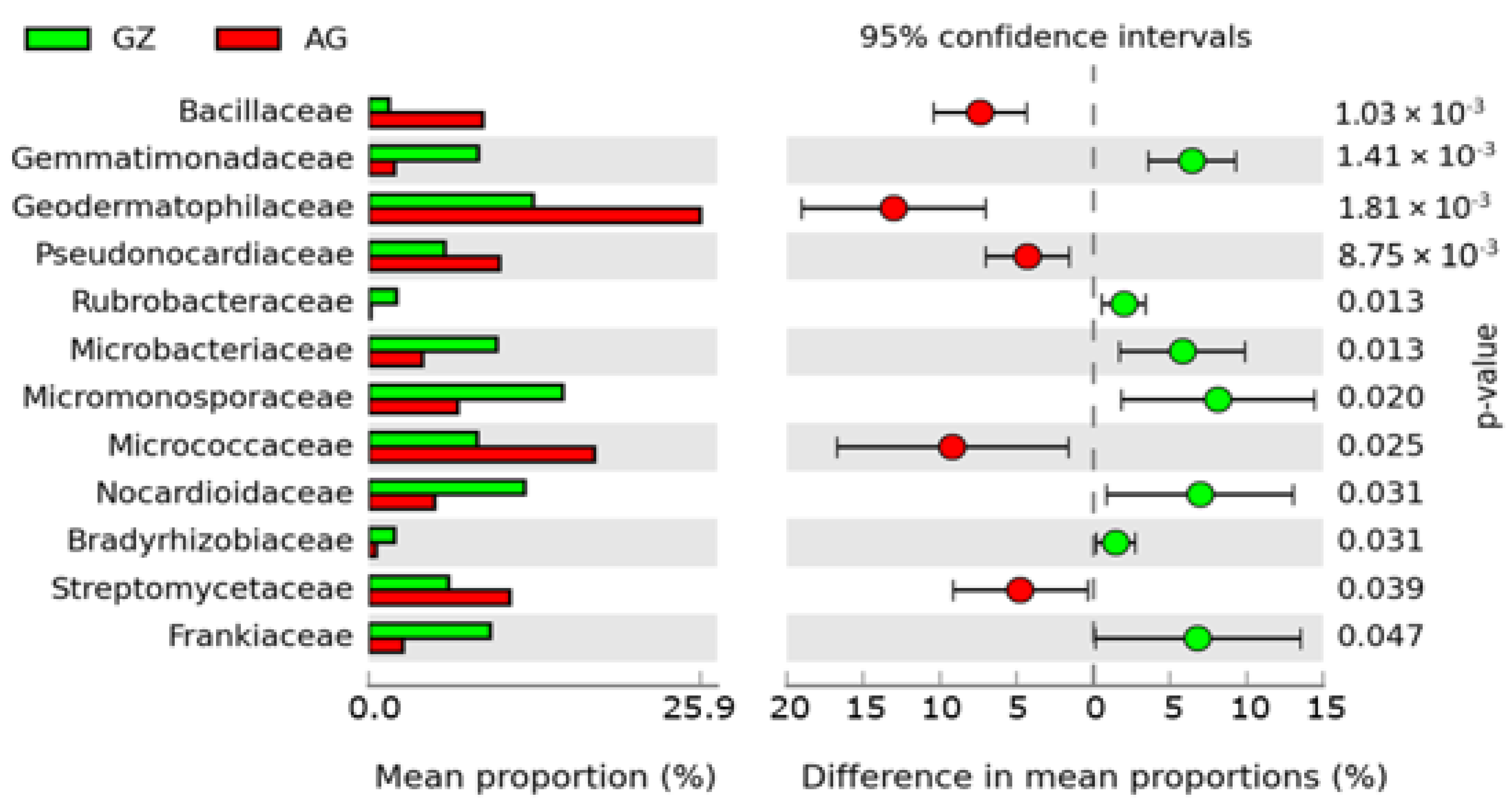
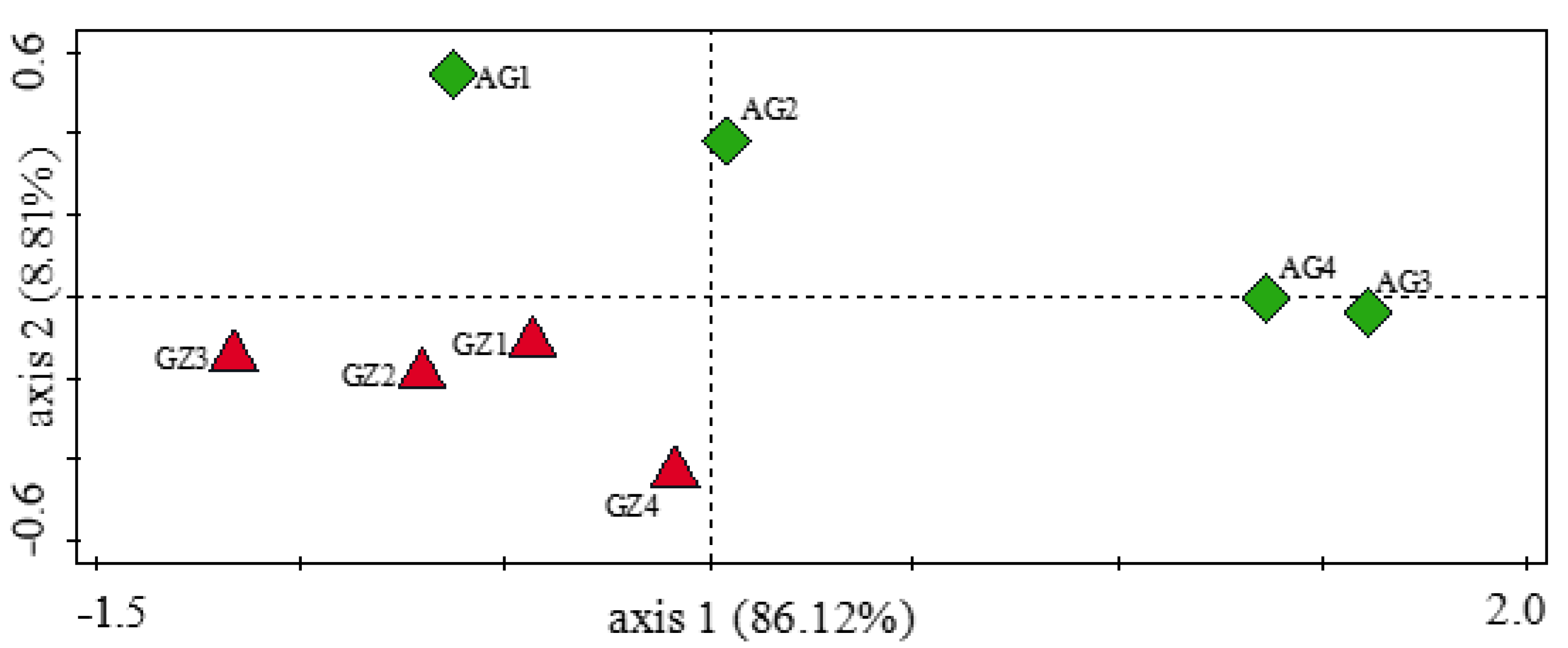
3.3. The Distribution of Microbes across the Maize Rhizosphere Soils
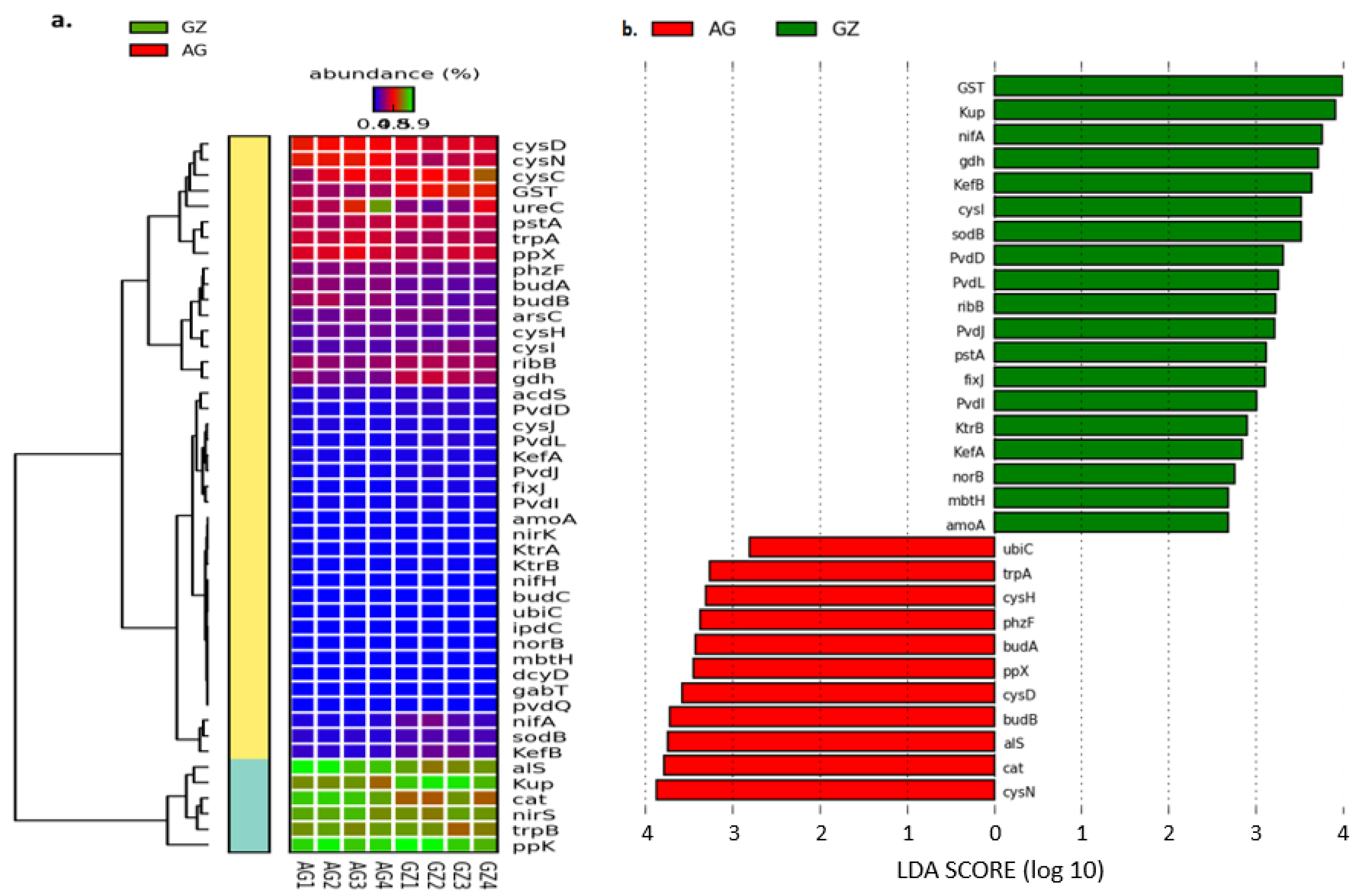
3.4. Microbial Genes That Enhance Plant Growth and Fitness Observed in the Maize Fields
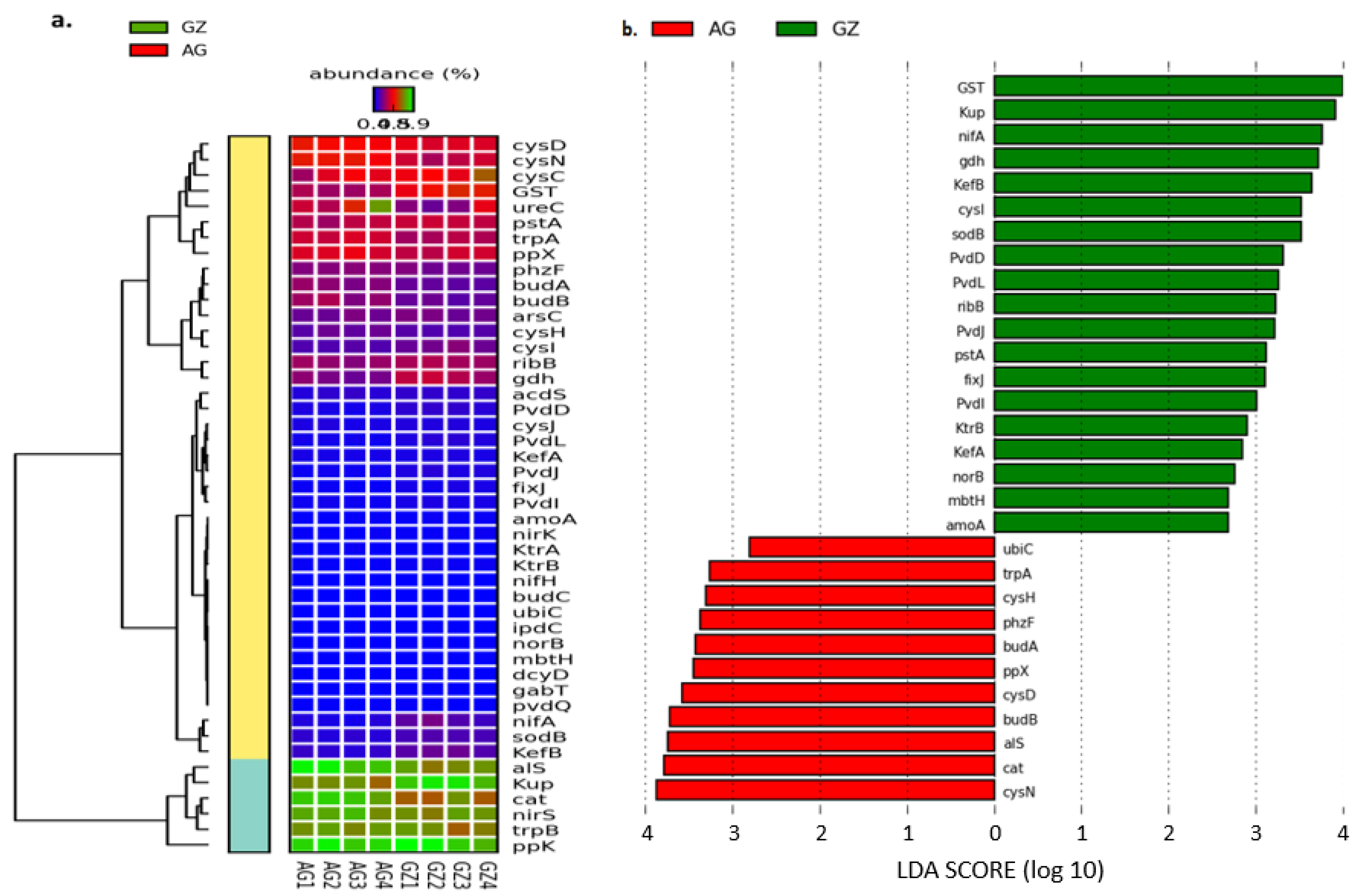
3.4.1. Genes That Facilitate Nutrient Mobilization and Plant Growth
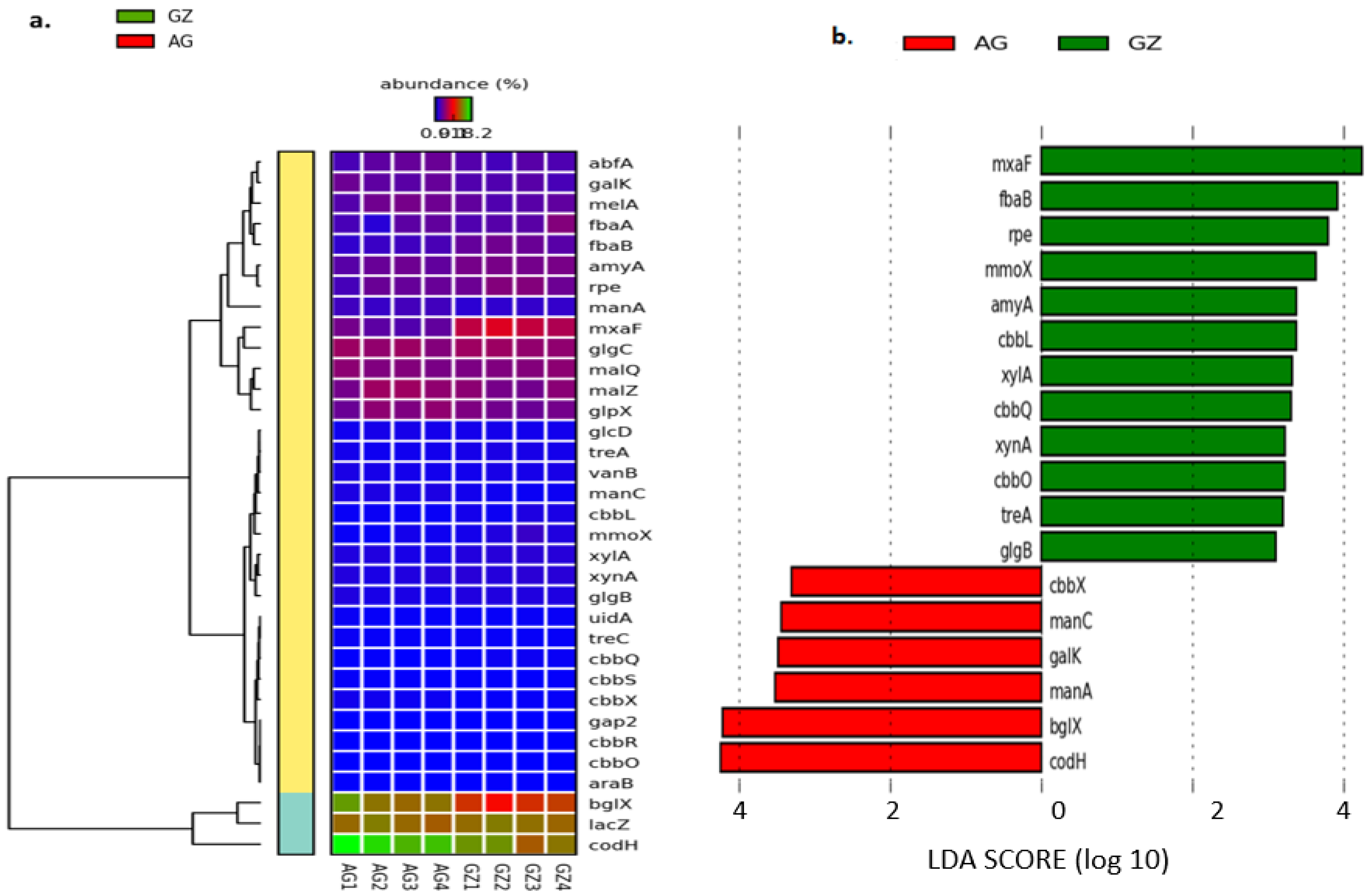
3.4.2. Microbial Genes Involved in Carbon Cycling
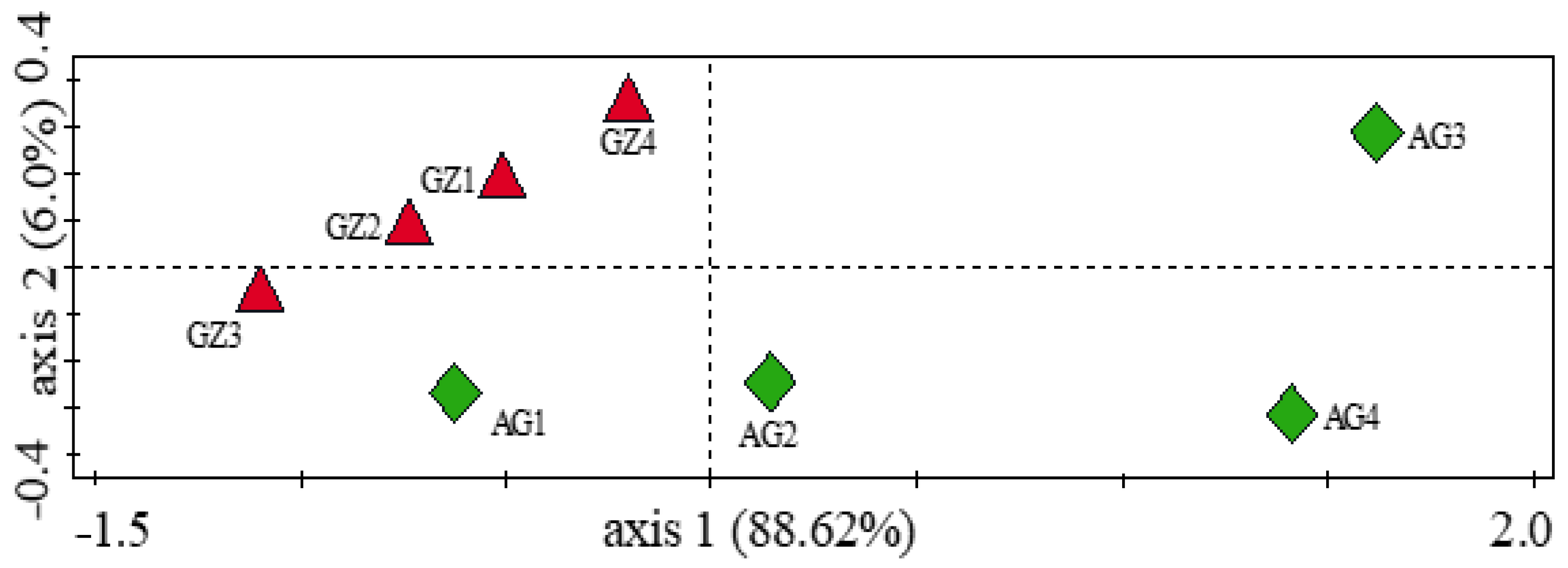
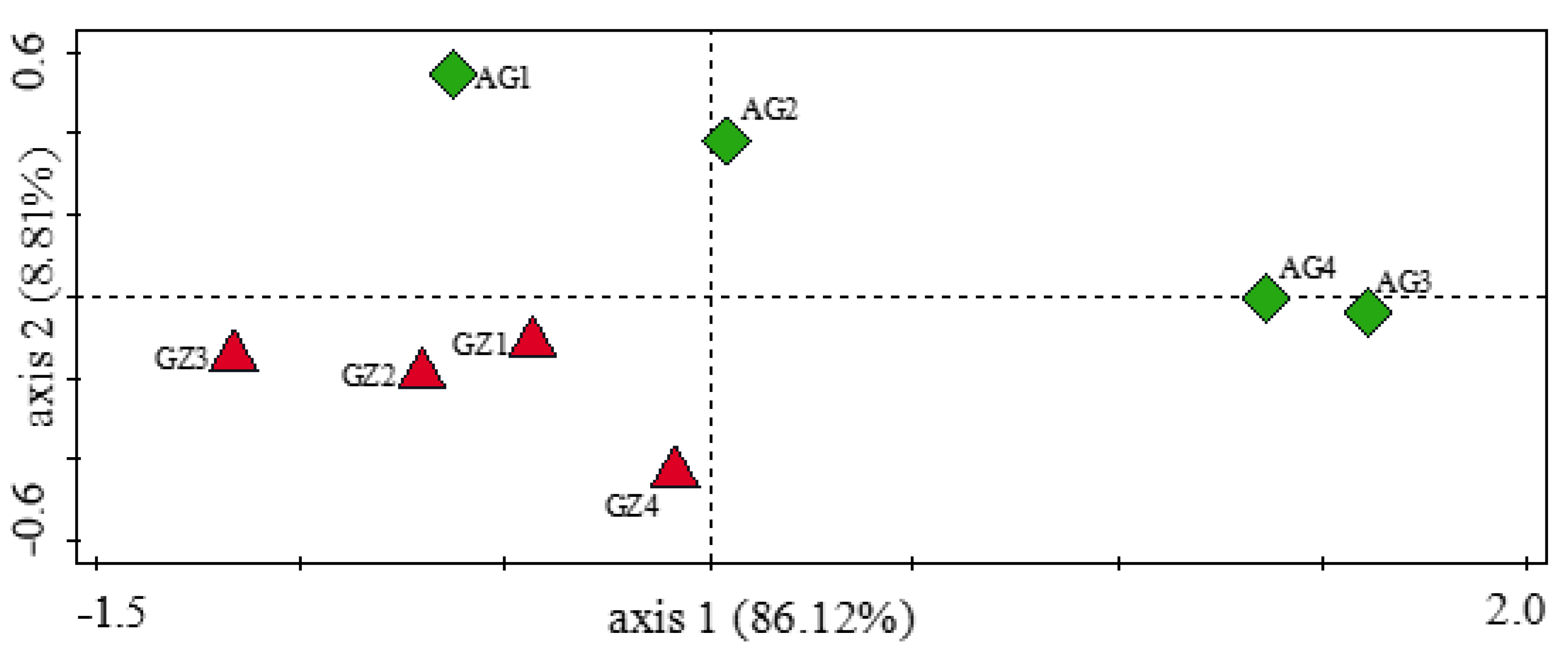
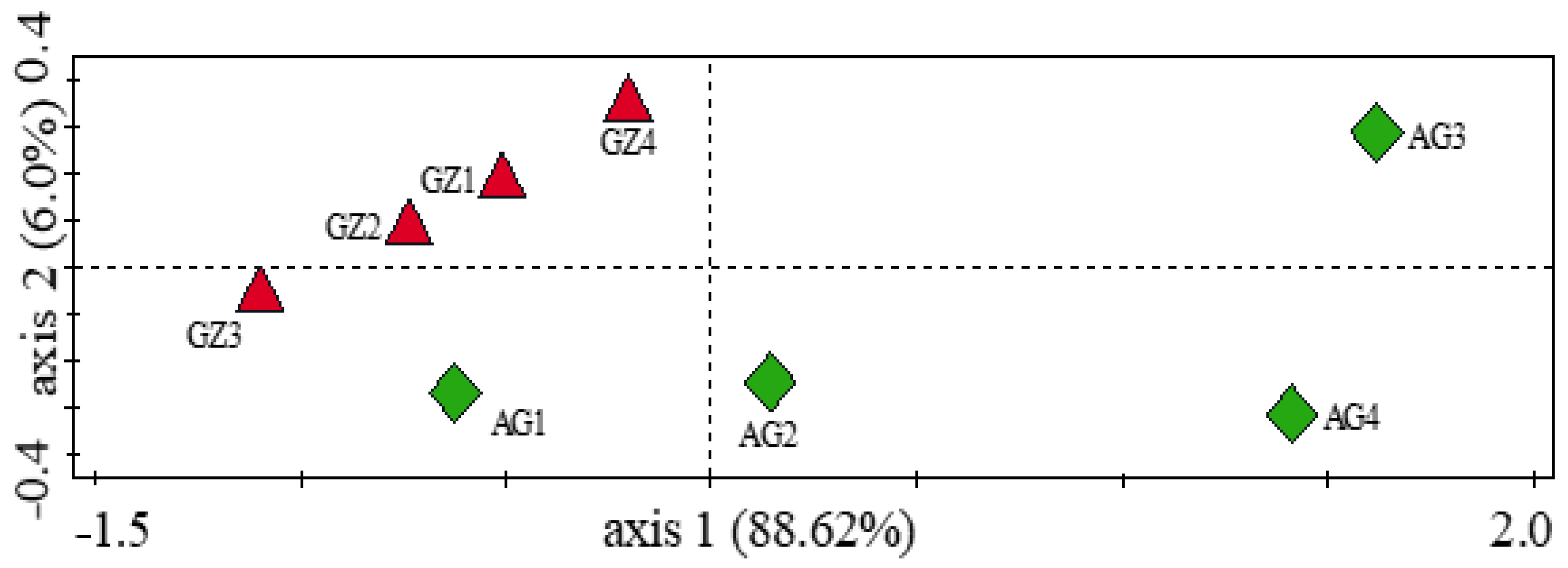
3.5. Influence of Soil Physicochemical Properties on the Diversity of Carbon-Cycling and Plant Growth-Promoting Genes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Manoharan, L.; Kushwaha, S.K.; Ahrén, D.; Hedlund, K. Agricultural land use determines functional genetic diversity of soil microbial communities. Soil Biol. Biochem. 2017, 115, 423–432. [Google Scholar] [CrossRef]
- Palansooriya, K.N.; Wong, J.T.F.; Hashimoto, Y.; Huang, L.; Rinklebe, J.; Chang, S.X.; Bolan, N.; Wang, H.; Ok, Y.S. Response of microbial communities to biochar-amended soils: A critical review. Biochar 2019, 1, 3–22. [Google Scholar] [CrossRef] [Green Version]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Cole, B.J.; Feltcher, M.E.; Waters, R.J.; Wetmore, K.M.; Mucyn, T.S.; Ryan, E.M.; Wang, G.; Ul-Hasan, S.; McDonald, M.; Yoshikuni, Y. Genome-wide identification of bacterial plant colonization genes. PLoS Biol. 2017, 15, e2002860. [Google Scholar] [CrossRef] [PubMed]
- Lebeis, S.L.; Paredes, S.H.; Lundberg, D.S.; Breakfield, N.; Gehring, J.; McDonald, M.; Malfatti, S.; Del Rio, T.G.; Jones, C.D.; Tringe, S.G. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef] [Green Version]
- Sloan, S.S.; Lebeis, S.L. Exercising influence: Distinct biotic interactions shape root microbiomes. Curr. Opin. Plant Biol. 2015, 26, 32–36. [Google Scholar] [CrossRef]
- König, S.; Vogel, H.-J.; Harms, H.; Worrich, A. Physical, chemical and biological effects on soil bacterial dynamics in microscale models. Front. Ecol. Evol. 2020, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Sui, X.; Zhang, R.; Frey, B.; Yang, L.; Li, M.-H.; Ni, H. Land use change effects on diversity of soil bacterial, Acidobacterial and fungal communities in wetlands of the Sanjiang Plain, northeastern China. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Berkelmann, D.; Schneider, D.; Meryandini, A.; Daniel, R. Unravelling the effects of tropical land use conversion on the soil microbiome. Environ. Microbiome 2020, 15, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, P.; Delgado-Baquerizo, M.; Jeffries, T.C.; Trivedi, C.; Anderson, I.C.; Lai, K.; McNee, M.; Flower, K.; Pal Singh, B.; Minkey, D. Soil aggregation and associated microbial communities modify the impact of agricultural management on carbon content. Environ. Microbiol. 2017, 19, 3070–3086. [Google Scholar] [CrossRef]
- Wagg, C.; Schlaeppi, K.; Banerjee, S.; Kuramae, E.E.; van der Heijden, M.G. Fungal-bacterial diversity and microbiome complexity predict ecosystem functioning. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Franco, N.; Martínez-Mena, M.; Goberna, M.; Albaladejo, J. Changes in soil aggregation and microbial community structure control carbon sequestration after afforestation of semiarid shrublands. Soil Biol. Biochem. 2015, 87, 110–121. [Google Scholar] [CrossRef]
- Wang, H.; Marshall, C.W.; Cheng, M.; Xu, H.; Li, H.; Yang, X.; Zheng, T. Changes in land use driven by urbanization impact nitrogen cycling and the microbial community composition in soils. Sci. Rep. 2017, 7, 1–12. [Google Scholar]
- Strickland, M.S.; Callaham Jr, M.A.; Gardiner, E.S.; Stanturf, J.A.; Leff, J.W.; Fierer, N.; Bradford, M.A. Response of soil microbial community composition and function to a bottomland forest restoration intensity gradient. Appl. Soil Ecol. 2017, 119, 317–326. [Google Scholar] [CrossRef] [Green Version]
- Dang, C.; Morrissey, E.M.; Neubauer, S.C.; Franklin, R.B. Novel microbial community composition and carbon biogeochemistry emerge over time following saltwater intrusion in wetlands. Glob. Chang. Biol. 2019, 25, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Barberán, A.; FERNÁNDEZ-GUERRA, A.; Bohannan, B.J.; Casamayor, E.O. Exploration of community traits as ecological markers in microbial metagenomes. Mol. Ecol. 2012, 21, 1909–1917. [Google Scholar] [CrossRef]
- Alami, M.M.; Xue, J.; Ma, Y.; Zhu, D.; Gong, Z.; Shu, S.; Wang, X. Structure, diversity, and composition of bacterial communities in rhizospheric soil of coptis chinensis franch under continuously cropped fields. Diversity 2020, 12, 57. [Google Scholar] [CrossRef] [Green Version]
- Fierer, N.; Barberán, A.; Laughlin, D.C. Seeing the forest for the genes: Using metagenomics to infer the aggregated traits of microbial communities. Front. Microbiol. 2014, 5, 614. [Google Scholar] [CrossRef]
- Schmidt, J.E.; Kent, A.D.; Brisson, V.L.; Gaudin, A.C. Agricultural management and plant selection interactively affect rhizosphere microbial community structure and nitrogen cycling. Microbiome 2019, 7, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Guo, Y.; Chen, X.; Wu, Y.; Zhang, L.; Cheng, J.; Wei, G.; Lin, Y. Natural revegetation of a semiarid habitat alters taxonomic and functional diversity of soil microbial communities. Sci. Total Environ. 2018, 635, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Raphael, J.P.; Calonego, J.C.; Milori, D.M.B.; Rosolem, C.A. Soil organic matter in crop rotations under no-till. Soil Tillage Res. 2016, 155, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.; Imran Ghani, M.; Li, Y.; Ding, H.; Meng, H.; Cheng, Z. Hiseq Base Molecular Characterization of Soil Microbial Community, Diversity Structure, and Predictive Functional Profiling in Continuous Cucumber Planted Soil Affected by Diverse Cropping Systems in an Intensive Greenhouse Region of Northern China. Int. J. Mol. Sci. 2019, 20, 2619. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Gao, Y.; Wang, S.; Xu, D.; Yu, H.; Wu, L.; Lin, Q.; Hu, Y.; Li, X.; He, Z. The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J. 2014, 8, 430–440. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Zhang, Y.; Zhang, P.; Trivedi, P.; Riera, N.; Wang, Y.; Liu, X.; Fan, G.; Tang, J.; Coletta-Filho, H.D. The structure and function of the global citrus rhizosphere microbiome. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Akinola, S.A.; Ayangbenro, A.S.; Babalola, O.O. The diverse functional genes of maize rhizosphere microbiota assessed using shotgun metagenomics. J. Sci. Food Agric. 2020. [Google Scholar]
- Enebe, M.C.; Babalola, O.O. Metagenomics Assessment of Soil Fertilization on the Chemotaxis and Disease Suppressive Genes Abundance in the Maize Rhizosphere. Genes 2021, 12, 535. [Google Scholar] [CrossRef] [PubMed]
- Ke, X.; Feng, S.; Wang, J.; Lu, W.; Zhang, W.; Chen, M.; Lin, M. Effect of inoculation with nitrogen-fixing bacterium Pseudomonas stutzeri A1501 on maize plant growth and the microbiome indigenous to the rhizosphere. Syst. Appl. Microbiol. 2019, 42, 248–260. [Google Scholar] [CrossRef]
- Paula, F.S.; Rodrigues, J.L.; Zhou, J.; Wu, L.; Mueller, R.C.; Mirza, B.S.; Bohannan, B.J.; Nüsslein, K.; Deng, Y.; Tiedje, J.M. Land use change alters functional gene diversity, composition and abundance in Amazon forest soil microbial communities. Mol. Ecol. 2014, 23, 2988–2999. [Google Scholar] [CrossRef] [PubMed]
- Muwawa, E.M.; Makonde, H.M.; Budambula, N.; Osiemo, Z.L. Chemical properties associated with guts, soil and nest materials of Odontotermes and Macrotermes species from Kenya. J. Biol. Environ. Sci. 2010, 4, 253–263. [Google Scholar]
- Bray, R.H.; Kurtz, L. Determination of total, organic, and available forms of phosphorus in soils. Soil Sci. 1945, 59, 39–46. [Google Scholar] [CrossRef]
- Walkley, A.; Black, I.A. An examination of the Degtjareff method for determining soil organic matter, and a proposed modification of the chromic acid titration method. Soil Sci. 1934, 37, 29–38. [Google Scholar] [CrossRef]
- Shi, J.-Y.; Yuan, X.-F.; Lin, H.-R.; Yang, Y.-Q.; Li, Z.-Y. Differences in soil properties and bacterial communities between the rhizosphere and bulk soil and among different production areas of the medicinal plant Fritillaria thunbergii. Int. J. Mol. Sci. 2011, 12, 3770–3785. [Google Scholar] [CrossRef]
- Nelson, D.W.; Sommers, L.E. Total carbon, organic carbon, and organic matter. Methods Soil Anal. Part 3 Chem. Methods 1996, 5, 961–1010. [Google Scholar]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A. The metagenomics RAST server–a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J. BLAT—the BLAST-like alignment tool. Genome Res. 2002, 12, 656–664. [Google Scholar] [CrossRef] [Green Version]
- Wilke, A.; Harrison, T.; Wilkening, J.; Field, D.; Glass, E.M.; Kyrpides, N.; Mavrommatis, K.; Meyer, F. The M5nr: A novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinform. 2012, 13, 141. [Google Scholar] [CrossRef] [Green Version]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, D206–D214. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [Green Version]
- Hammer, Ø.; Harper, D.A.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chukwuneme, C.F.; Ayangbenro, A.S.; Babalola, O.O.; Kutu, F.R. Functional diversity of microbial communities in two contrasting maize rhizosphere soils. Rhizosphere 2021, 17, 100282. [Google Scholar] [CrossRef]
- Yeager, C.M.; Dunbar, J.; Hesse, C.N.; Daligault, H.; Kuske, C.R. Polysaccharide degradation capability of Actinomycetales soil isolates from a semiarid grassland of the Colorado Plateau. Appl. Environ. Microbiol. 2017, 83, e03020-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamedi, J.; Mohammadipanah, F. Biotechnological application and taxonomical distribution of plant growth promoting actinobacteria. J. Ind. Microbiol. Biotechnol. 2015, 42, 157–171. [Google Scholar] [CrossRef] [PubMed]
- Tsegaye, Z.; Assefa, F.; Beyene, D. Properties and application of plant growth promoting rhizobacteria. Int. J. Curr. Trends Pharm. Med Sci. 2017, 2, 30–43. [Google Scholar]
- Chukwuneme, C.F.; Babalola, O.O.; Kutu, F.R.; Ojuederie, O.B. Characterization of actinomycetes isolates for plant growth promoting traits and their effects on drought tolerance in maize. J. Plant Interact. 2020, 15, 93–105. [Google Scholar] [CrossRef]
- Hartman, K.; van der Heijden, M.G.; Wittwer, R.A.; Banerjee, S.; Walser, J.-C.; Schlaeppi, K. Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 2018, 6, 1–14. [Google Scholar]
- Pérez-Jaramillo, J.E.; de Hollander, M.; Ramírez, C.A.; Mendes, R.; Raaijmakers, J.M.; Carrión, V.J. Deciphering rhizosphere microbiome assembly of wild and modern common bean (Phaseolus vulgaris) in native and agricultural soils from Colombia. Microbiome 2019, 7, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; Tang, Y.; Wang, Z.; Gou, Y.; Wang, J. Nitrogen-cycling genes and rhizosphere microbial community with reduced nitrogen application in maize/soybean strip intercropping. Nutr. Cycl. Agroecosystems 2019, 113, 35–49. [Google Scholar] [CrossRef]
- Valdés, M.; Pérez, N.-O.; Estrada-de Los Santos, P.; Caballero-Mellado, J.; Pena-Cabriales, J.J.; Normand, P.; Hirsch, A.M. Non-Frankia actinomycetes isolated from surface-sterilized roots of Casuarina equisetifolia fix nitrogen. Appl. Environ. Microbiol. 2005, 71, 460–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Tarabily, K.A.; Nassar, A.H.; Sivasithamparam, K. Promotion of growth of bean (Phaseolus vulgaris L.) in a calcareous soil by a phosphate-solubilizing, rhizosphere-competent isolate of Micromonospora endolithica. Appl. Soil Ecol. 2008, 39, 161–171. [Google Scholar] [CrossRef]
- Corre-Hellou, G.; Brisson, N.; Launay, M.; Fustec, J.; Crozat, Y. Effect of root depth penetration on soil nitrogen competitive interactions and dry matter production in pea–barley intercrops given different soil nitrogen supplies. Field Crop. Res. 2007, 103, 76–85. [Google Scholar] [CrossRef]
- Smith, C.R.; Blair, P.L.; Boyd, C.; Cody, B.; Hazel, A.; Hedrick, A.; Kathuria, H.; Khurana, P.; Kramer, B.; Muterspaw, K. Microbial community responses to soil tillage and crop rotation in a corn/soybean agroecosystem. Ecol. Evol. 2016, 6, 8075–8084. [Google Scholar] [CrossRef]
- Hu, J.; Jin, V.L.; Konkel, J.Y.; Schaeffer, S.M.; Schneider, L.G.; DeBruyn, J.M. Soil Health Management Enhances Microbial Nitrogen Cycling Capacity and Activity. Msphere 2021, 6, e01237-20. [Google Scholar] [CrossRef]
- Coskun, D.; Britto, D.T.; Shi, W.; Kronzucker, H.J. Nitrogen transformations in modern agriculture and the role of biological nitrification inhibition. Nat. Plants 2017, 3, 1–10. [Google Scholar] [CrossRef]
- Chowdhury, F.T.; Zaman, N.R.; Islam, M.R.; Khan, H. Anti-fungal secondary metabolites and hydrolytic enzymes from rhizospheric bacteria in crop protection: A review. J. Bangladesh Acad. Sci. 2020, 44, 69–84. [Google Scholar] [CrossRef]
- Igiehon, N.O.; Babalola, O.O. Rhizosphere microbiome modulators: Contributions of nitrogen fixing bacteria towards sustainable agriculture. Int. J. Environ. Res. Public Health 2018, 15, 574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.; Balser, T.C. Warming and nitrogen deposition lessen microbial residue contribution to soil carbon pool. Nat. Commun. 2012, 3, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enebe, M.C.; Babalola, O.O. Soil fertilization affects the abundance and distribution of carbon and nitrogen cycling genes in the maize rhizosphere. AMB Express 2021, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Mhete, M.; Eze, P.N.; Rahube, T.O.; Akinyemi, F.O. Soil properties influence bacterial abundance and diversity under different land-use regimes in semi-arid environments. Sci. Afr. 2020, 7, e00246. [Google Scholar] [CrossRef]
- Zhang, Q.; Wu, J.; Yang, F.; Lei, Y.; Zhang, Q.; Cheng, X. Alterations in soil microbial community composition and biomass following agricultural land use change. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Moebius-Clune, B.; Van Es, H.; Idowu, O.; Schindelbeck, R.; Kimetu, J.; Ngoze, S.; Lehmann, J.; Kinyangi, J. Long-term soil quality degradation along a cultivation chronosequence in western Kenya. Agric. Ecosyst. Environ. 2011, 141, 86–99. [Google Scholar] [CrossRef]
- Fujisao, K.; Khanthavong, P.; Oudthachit, S.; Matsumoto, N.; Homma, K.; Asai, H.; Shiraiwa, T. Impacts of the continuous maize cultivation on soil properties in Sainyabuli province, Laos. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kopittke, P.M.; Menzies, N.W.; Wang, P.; McKenna, B.A.; Lombi, E. Soil and the intensification of agriculture for global food security. Environ. Int. 2019, 132, 105078. [Google Scholar] [CrossRef] [PubMed]
- Barančíková, G.; Makovníková, J.; Halas, J. Effect of land use change on soil organic carbon. Agriculture (Pol’nohospodárstvo) 2016, 62, 10–18. [Google Scholar] [CrossRef]
- Kopittke, P.M.; Dalal, R.C.; Finn, D.; Menzies, N.W. Global changes in soil stocks of carbon, nitrogen, phosphorus, and sulphur as influenced by long-term agricultural production. Glob. Chang. Biol. 2017, 23, 2509–2519. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Ryan, J. 2015, 1. [CrossRef]
- Li, J.; Wan, X.; Liu, X.; Chen, Y.; Slaughter, L.C.; Weindorf, D.C.; Dong, Y. Changes in soil physical and chemical characteristics in intensively cultivated greenhouse vegetable fields in North China. Soil Tillage Res. 2019, 195, 104366. [Google Scholar] [CrossRef]
- Wang, X.; Tang, C. The role of rhizosphere pH in regulating the rhizosphere priming effect and implications for the availability of soil-derived nitrogen to plants. Ann. Bot. 2018, 121, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Hariharan, J.; Grewal, P.S.; Dick, W.A. Bacterial community dissimilarity in soils is driven by long-term land-use practices. Agrosystemsgeosciences Environ. 2020, 3, e20031. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Ren, L.; Zhang, J.; Luo, L.; Qin, P.; Zhou, Y.; Huang, C.; Tang, J.; Huang, H.; Chen, A. Population characteristics and influential factors of nitrogen cycling functional genes in heavy metal contaminated soil remediated by biochar and compost. Sci. Total Environ. 2019, 651, 2166–2174. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Pair | Ov. Avg. Dissimilarity | Contribution % of Plant Growth Promoting Genes | Ov. Avg. Dissimilarity | Contribution % of Carbon-Cycling Genes | ||||
|---|---|---|---|---|---|---|---|---|
| kup | ppk | trpB | codH | lacZ | mxaF | |||
| GZ and AG | 28.00 | 8.70 | 7.67 | 5.76 | 26 | 12.43 | 11.93 | 10.4 |
| GZ1 and GZ2 | 6.55 | 11.90 | 6.94 | 4.57 | 6.90 | 10.18 | 13.90 | 14.01 |
| GZ1 and GZ3 | 18.08 | 9.87 | 6.05 | 3.60 | 15.96 | 6.72 | 13.35 | 7.40 |
| GZ1 and GZ4 | 9.11 | 6.76 | 12.85 | 7.52 | 10.27 | 16.82 | 11.85 | 8.04 |
| GZ2 and GZ3 | 12.81 | 8.24 | 5.20 | 2.91 | 10.83 | 3.96 | 11.29 | 2.85 |
| GZ2 and GZ4 | 13.78 | 10.16 | 11.46 | 6.97 | 14.60 | 16.37 | 15.00 | 12.50 |
| GZ3 and GZ4 | 24.20 | 9.80 | 8.67 | 5.12 | 24.15 | 10.70 | 13.64 | 8.08 |
| AG1 and AG2 | 19.03 | 6.91 | 7.06 | 6.27 | 15.84 | 21.3 | 10.85 | 6.44 |
| AG1 and AG3 | 48.0 | 6.83 | 8.29 | 7.02 | 45.58 | 19.82 | 12.86 | 4.93 |
| AG1 and AG4 | 44.32 | 7.33 | 8.11 | 6.81 | 40.13 | 19.84 | 13.24 | 4.67 |
| AG2 and AG3 | 31.93 | 6.74 | 9.47 | 7.74 | 32.71 | 18.18 | 14.05 | 3.65 |
| AG2 and AG4 | 28.87 | 6.74 | 9.48 | 7.74 | 27.03 | 17.64 | 14.73 | 2.91 |
| AG3 and AG4 | 6.00 | 1.72 | 9.00 | 8.53 | 7.12 | 18.40 | 8.21 | 7.16 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chukwuneme, C.F.; Ayangbenro, A.S.; Babalola, O.O. Metagenomic Analyses of Plant Growth-Promoting and Carbon-Cycling Genes in Maize Rhizosphere Soils with Distinct Land-Use and Management Histories. Genes 2021, 12, 1431. https://doi.org/10.3390/genes12091431
Chukwuneme CF, Ayangbenro AS, Babalola OO. Metagenomic Analyses of Plant Growth-Promoting and Carbon-Cycling Genes in Maize Rhizosphere Soils with Distinct Land-Use and Management Histories. Genes. 2021; 12(9):1431. https://doi.org/10.3390/genes12091431
Chicago/Turabian StyleChukwuneme, Chinenyenwa Fortune, Ayansina Segun Ayangbenro, and Olubukola Oluranti Babalola. 2021. "Metagenomic Analyses of Plant Growth-Promoting and Carbon-Cycling Genes in Maize Rhizosphere Soils with Distinct Land-Use and Management Histories" Genes 12, no. 9: 1431. https://doi.org/10.3390/genes12091431
APA StyleChukwuneme, C. F., Ayangbenro, A. S., & Babalola, O. O. (2021). Metagenomic Analyses of Plant Growth-Promoting and Carbon-Cycling Genes in Maize Rhizosphere Soils with Distinct Land-Use and Management Histories. Genes, 12(9), 1431. https://doi.org/10.3390/genes12091431