Abstract
The evolution of the karyotype and genome size was examined in species of Crepis sensu lato. The phylogenetic relationships, inferred from the plastid and nrITS DNA sequences, were used as a framework to infer the patterns of karyotype evolution. Five different base chromosome numbers (x = 3, 4, 5, 6, and 11) were observed. A phylogenetic analysis of the evolution of the chromosome numbers allowed the inference of x = 6 as the ancestral state and the descending dysploidy as the major direction of the chromosome base number evolution. The derived base chromosome numbers (x = 5, 4, and 3) were found to have originated independently and recurrently in the different lineages of the genus. A few independent events of increases in karyotype asymmetry were inferred to have accompanied the karyotype evolution in Crepis. The genome sizes of 33 Crepis species differed seven-fold and the ancestral genome size was reconstructed to be 1C = 3.44 pg. Both decreases and increases in the genome size were inferred to have occurred within and between the lineages. The data suggest that, in addition to dysploidy, the amplification/elimination of various repetitive DNAs was likely involved in the genome and taxa differentiation in the genus.
1. Introduction
Chromosomal changes, both numerical and structural, are acknowledged to be important mechanisms that accompany speciation and diversification in plants [1]. The large variation in chromosome numbers in the plant kingdom is the result of two major mechanisms—polyploidy and dysploidy [2]. Polyploidisation (whole genome duplication) and subsequent diploidisation seem to play a greater role in the evolution of angiosperms than in other eukaryotes [3]. There is evidence that most, if not all, angiosperms are ancient polyploids [4]. Dysploidy involves various types of structural rearrangements of chromosomes, which often result in decreases or increases in chromosome numbers [5,6]. The chromosome morphology can also be altered by the amplification/loss of DNA sequences [7]. Accumulation and/or loss of DNA sequences (mainly repetitive DNA) are mechanisms that lead to changes in the total karyotype length and genome size [8,9].
Crepis sensu lato (s.l.), which consists of approximately 200 annual and perennial species, is one of the largest genera in Asteraceae [10]. Most Crepis species are diploid with relatively low numbers of rather large and well-differentiated chromosomes. Several chromosome base numbers (x = 3, 4, 5, 6, and 11 [11]) and significant differences in genome size from 0.72 pg/1C to 32.75 pg/1C [12] have been reported in this genus previously. Thus, Crepis has been an excellent system for investigating the evolution of the karyotype [13,14]. The first cytogenetic and taxonomical study of numerous Crepis species defined several karyomorphotypes based on the number and morphology of the chromosomes and the symmetry/asymmetry of the karyotypes [15]. The first comprehensive sectional classification of Crepis was largely based on a combination of chromosomal and morphological characters of the species [11,15,16]. Thus, the genus Crepis has been considered to be the first model plant group in which chromosomal evolution plays an important role in speciation (e.g., [13,17,18,19]). The morphological, cytological and physiological evidence suggested that a decrease in the base chromosome numbers and a shortening of the life cycle accompanied the evolution of the genus [20]. Decreases in the chromosome numbers from an ancestral 2n = 12 to the derived 2n = 10, 8, and 6 have been suggested to have occurred mostly via the reciprocal translocations between non-homologous chromosomes [20]. In the diversification and speciation of the genus Crepis, the decreases in the base chromosome numbers and increases in karyotype asymmetry were suggested to be the main direction of the evolution of the karyotype [13,20].
Symmetrical karyotypes composed of mostly metacentric and submetacentric chromosomes of similar sizes were usually considered to be plesiomorphic. Asymmetrical karyotypes, considered to be apomorphic, are composed of highly variable in size chromosomes. Most of their chromosomes have terminally or subterminally localised centromeres [13,21]. This suggested that the most derived Crepis species possessed x = 3 chromosomes, asymmetrical karyotypes that were primarily annuals [13,20].
Molecular phylogenetic studies of the genus Crepis sensu lato (s.l.) tested Babcock’s hypothesis in a phylogenetic framework [22] and proposed a new classification of this genus. Three evolutionary lineages were distinguished based on the analyses of 75 taxa: (i) species with a chromosome base number x = 7, corresponding to Babcock’s section Ixeridopsis and now placed in genus Askellia; (ii) Babcock’s sections Intybellia, Lagoseris, Phaecasium, Microcephalum, and Pterotheca, as well as two other genera, Lapsana and Rhagadiolus, now included in the Lagoseris evolutionary lineage; and (iii) Crepis sensu stricto (Crepis s.s.), which comprised the remaining analysed Crepis species [22,23]. The new sections were highly heterogeneous compared to Babcock’s sections, also concerning base chromosome numbers, suggesting that the infrageneric classification of Babcock did not represent natural groups [20,22,23].
Modern cytogenetic methods combined with molecular phylogenetic analyses greatly facilitate the analyses of the trends in chromosomal evolution that accompany and/or follow diversification and speciation in plants. The aim of the study was to analyse the patterns of chromosome number and genome size evolution in Crepis s.l. species. Chromosome numbers, karyotype structure, genome size, and DNA sequence information were obtained de novo for Crepis species and Lapsana communis included in this study. All of the analyses were performed on the same set of species and individuals, which enabled the perfect correlation of the molecular and cytogenetic data.
2. Materials and Methods
2.1. Plant Material
Fifty-five accessions of 45 species of Crepis representing two evolutionary lineages Lagoseris and Crepis s.s. as well as Lapsana communis, which, according to Enke et al. [23], belongs to the Lagoseris evolutionary lineage, were analysed (Table 1). The analysed samples represented about a quarter of all currently recognized Crepis species [20]. Picris hieracioides, Lactuca serriola, and Sonchus oleraceus were used as outgroups for phylogenetic analyses. The plants were grown from seeds in the greenhouse of the University of Silesia in Katowice under a 16 h/8 h photoperiod at 19 ± 2 °C. The vouchers were deposited at the Herbarium KTU (University of Silesia, Chorzów, Poland; Table 1).

Table 1.
Species names, collection numbers, places of seed collecting, voucher numbers of the analysed taxa, and the GenBank accessions numbers of the sequences obtained in this study.
2.2. DNA Amplification and Sequencing
Total genomic DNA was isolated from fresh leaf tissue using a modified CTAB method [24]. Genomic DNA from each sample was checked for quality and quantity using an ND-1000 (peqLab, Erlangen, Germany). The PCR amplification of the nuclear rDNA ITS (internal transcribed spacer; nrITS) region was carried out according to Venora et al. [25] using the primers ITS4 and ITS5 [26]. Based on the analyses of Shaw et al. [27], the four non-coding plastid DNA spacer regions (rpl32-trnL, rps16-trnK, ndhC-trnV and psbD-trnT), which have a high number of potentially informative characters in euasterids II, were selected for the analyses. The cpDNA markers were amplified using the primers from Shaw et al. ([28]; Table S1). The PCR mixture for the amplification contained a 1x Color OptiTaq PCR Master Mix (EURx, Gdansk, Poland), 0.5 µM of each forward and reverse primer (Genomed, Warsaw, Poland), and 50 ng of the DNA template. The GeneAmpPCR System 9700 (Applied Biosystems, Waltham, MA, USA) and the conditions described in Blöch et al. [29] were used for the amplification. The PCR products were treated with E. coli Exonuclease I and FastAP Thermosensitive Alkaline Phosphatase (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions and the cycle sequencing was performed using a 3730xl DNA Analyzer (Applied Biosystems; USA) in a commercial facility (Genomed; Warsaw, Poland or Macrogen; Seoul, Korea). The template DNAs that were used for the nrITS and cpDNA amplifications were always derived from the same isolate. All of the sequences were deposited in GenBank (accession numbers in Table 1).
2.3. Sequence Alignment and Phylogenetic Analyses
Most of the phylogenetic analyses were performed using the DNA sequences of four concatenated cpDNA regions and the nrITS that was obtained in this study. Additionally, an extended nrITS dataset, which contained the sequences that were obtained in this study as well as previously published sequences were also analysed [12]. The sequences were assembled using DNA Baser version 3 (Heracle BioSoft S.R.L., Pitesti, Romania). Multiple sequence alignments were performed 20 times using webPRANK [30] and MergeAlign [31] was then used to obtain a consensus of the multiple sequence alignments. The phylogenetic relationships were inferred independently for the nrITS and for the four concatenated plastid regions using the maximum likelihood, as implemented in IQ-TREE version 0.9.5 [32]. The best model of sequence evolution for the ML analyses was determined using the Bayesian information criterion as implemented in IQ-TREE [33]. The best fit models were TIM3e + G4 for nrITS and TVM + F + G4 for the plastid data sets. The significance of the inferred relationships was assessed via bootstrapping with 1000 replicates. Picris hieracioides, Lactuca serriola, and Sonchus oleraceus were used as the outgroup taxa. The resulting phylogenetic trees were created using FigTree v.1.3.1 [34]. Bootstrap support values below 75 were excluded from the figures.
2.4. Chromosome Preparation and Karyotype Analyses
Young leaves were pretreated with 2 mM 8-hydroxyquinoline (Sigma, Burlington, MA, USA) for 2 h at room temperature and for 2 h at 4 °C, fixed in methanol:glacial acetic (3:1) and stored at −20 °C until use. The mitotic metaphase chromosome spreads were prepared according to Dydak et al. [35]. The chromosome preparations were stained with DAPI (4′,6-diamidino-2-phenylindole) and analysed under a Zeiss AxioImager.Z.2 fluorescent microscope (ZEISS, Germany). The karyotype analyses were performed on at least ten high-quality mitotic metaphase spreads per accession. The chromosome length was measured using ImageJ software ver. 1.50 [36]. The chromosomes of each analysed cell were arranged into the karyotypes by decreasing length. The chromosome nomenclature of Levan et al. [37] was used. The degree of karyotype asymmetry was estimated according to Paszko [38]. The asymmetry index is an indicator of the levels of the heterogeneity of chromosome length and the positions of the centromeres in a given karyotype. The asymmetry index (A1) was calculated using the following equation:
where CVCL is the relative variation in chromosome length; CVCL = (SCL is a standard deviation and XCL is the mean chromosome length), whereas CVCI is the relative variation centromeric index CVCI = (SCI is the standard deviation and XCI is the mean centromeric index).
2.5. Genome Size Measurements
The genome sizes were measured using flow cytometry. Brachypodium hybridum Catalán, Joch.Müll., Hasterok & G. Jenkins ABR113 (2C DNA = 1.265 pg; [39]); Solanum lycopersicum L., Stupicke’ (1.96 pg/2C DNA; [40]; Solanum pseudocapsicum L. (2.58 pg/2C DNA, [41]); Zea mays L. ‘CE-777’ (2C = 5.43 pg/2C DNA, [42]); Pisum sativum L. ‘Ctirad’ (9.09 pg/2C DNA, [43]); and Secale cereale subsp. cereale L. (16.01 pg/2C DNA, [43]) were used as the internal standards, depending on the genome sizes of the measured samples. The youngest fully developed leaves of the analysed Crepis species and the appropriate internal standard (the information about the standards were added to Table 2 in results section) were chopped together in a Petri dish in 500 μL of a nuclei extraction buffer using a razor blade (CyStain PI Precision P Sysmex 05-5022). The nuclei suspension was filtered through a 30 μm mesh (CellTrics, Sysmex, Kobe, Japan) and stained with a staining buffer containing propidium iodide, RNase (CyStain PI Sysmex Precision P 05-5022, Kobe, Japan) and 1% β-mercaptoethanol (Sigma, Burlington, MA, USA) according to the manufacturer’s instructions. The samples were then analysed using a flow cytometer (CyFlow Space, Sysmex, Kobe, Japan) equipped with a 532 nm green laser. At least 10,000 nuclei were analysed for each sample. The sizes of the nuclear genomes were calculated as the linear relationship between the ratio of the 2C DNA peaks of a sample and the standard. Due to the high content of secondary metabolites, which prevented a flow cytometry analysis, the genome size could not be estimated for 12 species. The accepted CVs were less than 5%, except for C. kotschyana (5.78%) and diploid C. vesicaria (5.93%), for which measurements with CVs lower than 6% were accepted. The 1Cx-value and DNA content of one non-replicated monoploid genome with chromosome number x were calculated according to Greilhuber et al. [44].
Table 2.
Species names, karyotype formulas, asymmetry indices, and genome size of the analysed taxa.
2.6. Ancestral State Reconstructions
The phylograms that resulted from the ML analysis (branch length information included) were used to infer the evolution of the chromosome numbers, the karyotype asymmetry, and genome size. The analyses were performed using the better supported cpDNA phylogram. Previously published genome size values for Crepis (Tables S2 and S3) [12] were added to the newly obtained results and together mapped on the phylogram resulting from the ML analysis of the extended nrITS dataset. The maximum likelihood analyses were performed under the CONST_RATE model (for the nrITS data set) or the CONST_RATE_DEMI model (for the cpDNA dataset), as implemented in ChromEvol 2.0. software [45]. For the ChromEvol analyses, the best-fit model was tested using an AIC test (Table S4). For the best-fitted model, the analyses were rerun with parameters that were fixed to those that were optimised in the first run using 10,000 simulations to compute the expected number of changes along each branch as well as the ancestral haploid chromosome numbers at the nodes. Picris hieracioides, Sonchus oleraceus, and Lactuca serriola were used to root the tree. Bootstrap support values less than 75 were excluded. The chromosome base numbers for Crepis pontana, C. acuminata, C. atribarba, C. modocensis, and C. intermedia were obtained from the literature [20,46] because of problems with obtaining good-quality meristematic tissue for the chromosomal analyses. The base chromosome number for the polyploid C. biennis (x = 5) was adopted from [47]. The evolution of the asymmetry index and genome sizes were analysed using maximum likelihood as implemented in the package phytools in the R software [48].
3. Results
3.1. Phylogenetic Analysis
The phylogenetic relationships of the 45 Crepis species and Lapsana communis were inferred from analyses of their nrITS sequences and four plastid regions (rpl32-trnL, ndhC-trnV, rps16-trnK and trnT-psbD). A total of 61 nrITS and 244 cpDNA sequences (61 sequences per each of the four chloroplast markers) were analysed. Three species, Picris hieracioides, Lactuca serriola, and Sonchus oleraceus, were used as the outgroups. The length of the isolated nrITS regions among the analysed species ranged from 540 to 598 bp. The final alignment was 668 bp long (including gaps) with 241 Characters that were parsimony informative. Only one ribotype of the nrITS was amplified for all of the studied accessions. The total length of the analysed concatenated plastid DNA regions ranged from 2948 to 3577 bp (the data for the individual chloroplast markers are listed in Table S5). The concatenated alignment of the cpDNA was 5693 bp long (including gaps) with 720 characters that were parsimony informative.
Two major well-supported clades (nrITS BS100 and cpDNA BS100) corresponding to the evolutionary lineages of Lagoseris and Crepis s.s. (Figure 1) were consistently recovered in both the nrITS and cpDNA ML analyses. Crepis s.s., which contained most of the analysed species, was monophyletic in both of the nrITS and cpDNA analyses; however, the nrITS- and cpDNA-tree topologies differed in their branching patterns. The clades that were recovered in the cpDNA marker analyses were labelled with Roman numerals (clades I–IV) and the subclades with Roman numerals and lower-case letters (subclades IVa–IVc). The clades in the nrITS phylogenetic analyses were labelled with Arabic numerals (clades 1–4) and the subclades were labelled with Arabic numerals and lower-case letters (subclades 4a–4c). Four clades were distinguished in Crepis s.s. in all of the analyses. Clade 1 (BS81), which was recovered in the nrITS data analysis, included five perennial European species and the central Asian C. lyrata. This group of species was monophyletic in the nrITS phylogeny, but not in the cpDNA analysis in which the species were placed in two highly supported clades instead: clade I (BS99) and clade II (BS100; Figure 1). The second monophyletic clade 2 (BS96), which was recovered in the nrITS analysis, comprised nine, mostly annual Crepis species, with the exception of the perennial C. sibirica. These species were placed in subclade IVb (BS97) in the cpDNA tree together with C. setosa (Figure 1). Six European and/or Middle East perennial species, which were recovered in clade 3 (BS96) of the nrITS tree, were placed in subclade IVa (BS100) in the cpDNA phylogeny. Nearly half of the analysed Crepis s.s. species were placed in clade 4 in the nrITS phylogeny. Three highly supported subclades could be distinguished within this clade: (i) subclade 4a (BS90), which consisted of species that are native to North America; (ii) subclade 4b (BS86), which comprised C. biennis, C. setosa, and C. nicaeensis; and (iii) subclade 4c, which comprised both annual and perennial species from Europe and North Africa (from Algeria to Morocco). However, in the cpDNA phylogeny, the North American species together with C. tectorum, C. nigrescens, C. nicaeensis, and C. biennis formed the monophyletic clade III (BS100).
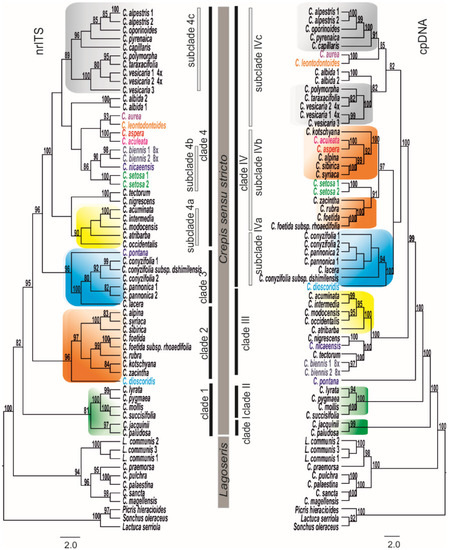
Figure 1.
Phylogenetic relationships among the analysed Crepis species based on the nrITS and the cpDNA data sets. Bootstrap support values are indicated at each node. Names of the species that were recovered in different positions in the two datasets are indicated in colour. The trees were rooted with Picris hieracioides, Lactuca serriola, and Sonchus oleraceus.
Three main evolutionary lineages were distinguished in the phylogenetic tree that resulted from the analysis of the newly obtained nrITS sequences and previously published data (species of Askellia. Lagoseris and Crepis s.s.; Figure S1; Table S3; [22]). Among the species of Crepis s.s., five main clades were identified, in congruence with earlier reports [22,23].
3.2. Chromosome Number
The chromosome numbers were analysed for 50 accessions (Table 2) representing 40 Crepis species and three accessions of L. communis. Most of the analysed Crepis species were diploids and only four polyploids were found, C. biennis (2n = 8x = 40), C. occidentalis (2n = 4x = 44), and C. vesicaria, which were represented by two polyploid accessions (both 2n = 4x = 16) and one diploid accession (2n = 2x = 8; Table 2; Figure S2). Five different chromosome base numbers were found among the diploid Crepis species: x = 3, 4, 5, 6, and 11 (Table 2; Figure 2, Figure 3 and Figure S4). The most common chromosome base numbers were x = 4 found in 20 species and x = 5 found in ten species. Six species had x = 6 and only two species had a chromosome base number of x = 3 (C. capillaris and C. zacintha). The chromosome base number of x = 11 was found in C. occidentalis, a representative of the American Crepis agamic complex (2n = 4x = 44; Figure S5). L. communis from the Lagoseris clade was diploid with 2n = 14.
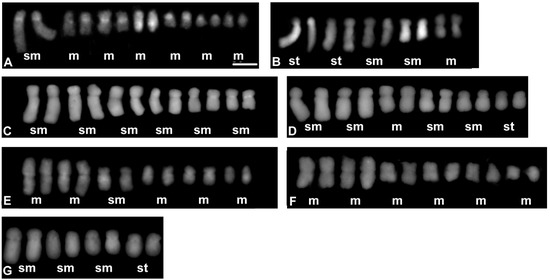
Figure 2.
Karyograms representing each of the karyotype formula distinguished among the analysed Crepis species: (A) Lapsana communis; (B) C. magellensis; (C) C. jacquinni; (D) C. paludosa; (E) C. succisifolia; (F) C. lyrate; (G) C. nicaeensis. Letters below each pair of chromosomes indicate the type of chromosome: m—metacentric; sm—submetacentric; and st—subtelocentric. The metaphase plates used to prepare the karyograms are presented in Figure S2. Scale bar = 5 µm.
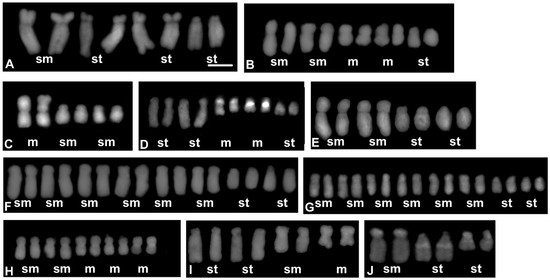
Figure 3.
Karyograms representing each of the karyotype formula distinguished among the analysed Crepis species: (A) C. pannonica; (B) C. foetida subsp. rhoaedifolia; (C) C. zacintha; (D) C. syriaca; (E) C. kotschyana; (F) C. vesicaria 2; (G) C. vesicaria 1; (H) C. leontodontoides; (I) C. oporinoides; (J) C. capillaris. Letters below each pair of chromosomes indicate the type of chromosome: m—metacentric; sm—submetacentric; and st—subtelocentric. The metaphase plates used to prepare the karyograms are presented in Figure S3. Scale bar = 5 µm.
The ML analysis (ChromEvol 2.0) was based on the cpDNA due to better support obtained for this marker. The ancestral chromosome base number for the common ancestor of Lagoseris and Crepis s.s. was inferred as x = 6 (pp = 0.97; Figure 4). The analysis of the cpDNA data sets suggested fourteen reductions (with expectation above 0.5) in the chromosome base numbers (Figure 4), three in the Lagoseris group and eleven in Crepis s.s. The ancestral base numbers for clades I and II was reconstructed as x = 6. The chromosome base number of x = 5 was inferred for the common ancestor of the species in clades III and IV (Figure 4). There was a further decrease in the chromosome base number from x = 5 to x = 4 in the evolutionary lineage of C. tectorum and C. nigrescens and in the evolutionary lineage of C. nicaeensis in clade III (Figure 4). A reduction in the chromosome base number from x = 5 to x = 4 accompanied the diversification of the common ancestor of the species in subclade IVa (Figure 4). Several events of descending dysploidy were also inferred for subclades IVb and IVc: (i) from x = 5 to x = 4 in the C. kotschyana lineage and C. setosa lineages; independently in the group that consisted of C. taraxacifolia, C. vesicaria, and C. polymorpha; in the lineages of C. aculeata and C. aspera; as well as for the common ancestor of the group, consisting of C. alpestris, C. oporinoides, C. pyrenaica, and C. capillaris; (ii) from x = 5 to x = 3 in C. zacintha; and (iii) from x = 4 to x = 3 in the C. capillaris lineage (Figure 4).
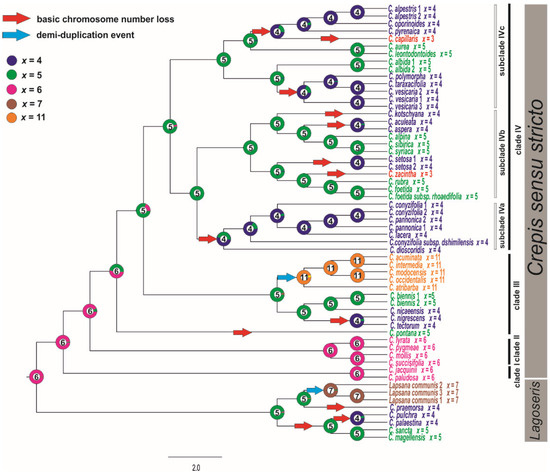
Figure 4.
Ancestral character state reconstruction of the chromosome base numbers of the analysed species of Crepis s.l. The chromosome base numbers have been mapped on the ML tree of concatenated cpDNA sequences using the maximum likelihood method implemented in ChromEvol 2.0 software [45]. The tree was rooted with Picris hieracioides, Lactuca serriola, and Sonchus oleraceus.
3.3. Karyotype Structure
A morphometric analysis of the chromosomes was performed for 47 accessions representing 38 Crepis species and three accessions of L. communis. The majority of the analysed species (38 accessions of 31 species) had symmetrical karyotypes (Table 2). Only nine of the analysed Crepis species had karyotypes with a higher asymmetry index (Table 2). The asymmetry index (AI) values were mapped onto the ML phylogenetic tree using the maximum likelihood (Figure 5 and Figure S6). The analysis was performed using better supported the cpDNA phylogenetic trees. The symmetrical karyotype was inferred as the ancestral state for the common ancestor of Lagoseris and Crepis s.s. (Figure 5). In the evolutionary lineage of Lagoseris, an increase in the karyotype asymmetry accompanied the speciation of C. praemorsa and L. communis. An increase in the karyotype asymmetry accompanied also the evolution of several Crepis s.s. species from subclades IVb in the cpDNA tree (C. alpina, C. sibirica, C. syriaca, C. rubra, and C. zacintha) and from subclade IVc (C. oporinoides, C. pyrenaica, and C. capillaris; Figure 5).
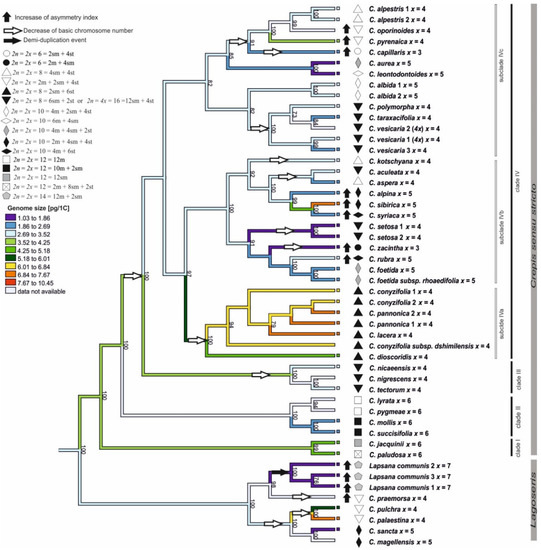
Figure 5.
Karyotype evolution in the analysed species of Crepis s.l. Genome sizes were mapped on the ML tree of concatenated cpDNA sequences using the maximum likelihood method implemented in Phytools. Karyotype formulas (symbols preceding the species names) and the increases in karyotype asymmetry (black arrows) are indicated for all species. The genome sizes, karyotype formulas, and asymmetry indexes of each species are presented in Table 2. The figure includes only species for which all three datatypes (chromosome number, karyotype formula, and asymmetry index) were provided. The tree was rooted with Picris hieracioides, Lactuca serriola, and Sonchus oleraceus.
Fourteen different karyotype formulas were found based on the morphometric analyses of Crepis chromosomes (Table 2 and Figure 2, Figure 3 and Figure S4). Four different karyotype formulas were observed in the species from clade I and clade II that had the same chromosome base number as the ancestral recovered state (x = 6). The karyotypes of the species from clade II mainly consisted of metacentric chromosomes (C. lyrata and C. pygmeae with the karyotype formula 2n = 2x = 12 = 12m; Figure 2F and Figure S2; C. succisifolia and C. mollis with the karyotype formula 2n = 2x = 12 = 10m + 2sm; Figure 2E and Figure S4; Table 2), whereas the karyotypes of the species from clade I mainly had submetacentric chromosomes (C. jacquinii with 2n = 2x = 12 = 12sm; and C. paludosa with 2n = 2x = 12 = 2m + 8sm + 2st; Figure 2C,D; Table 2).
Ten of the analysed species with a chromosome base number of x = 5 had five different karyotype formulas. The karyotypes of most of the species consisted of metacentric, submetacentric, and subtelocentric chromosomes (Figure 2, Figure 3 and Figure S4) and represented three different karyotype formulas: (i) 2n = 2x = 10 = 4m + 2sm + 4st (C. albida; Figure S4; Table 2); (ii) 2n = 2x = 10 = 2m + 4sm + 4st (C. magellensis, C. sibirica, C. alpina and C. sancta; Figure 2B and Figure S4; Table 2); and (iii) 2n = 2x = 10 = 4m + 4sm + 2st (C. aurea and C. foetida; Figure S4; Table 2). The karyotype of C. leontodontoides exclusively consisted of metacentric and submetacentric chromosomes (2n = 2x = 10 = 6m + 4sm; Figure 3H). Two other species (C. syriaca and C. rubra; Figure 3D and Figure S4; Table 2) had metacentric and subtelocentric chromosomes (2n = 2x = 10 = 4m + 6st). The species with x = 5 were mainly included in subclades IVb and IVc; however, even closely related species with the same chromosome number differed in their karyotype formulas (e.g., C. aurea and C. leontodontoides; Figure 5). Among the species with a chromosome base number of x = 4, four different karyotype formulas were distinguished. Species from clade III and the group of species that is closely related to C. vesicaria (from subclade IVc) had karyotypes with submetacentric and subtelocentric chromosomes (2n = 2x = 8 = 6sm + 2st or 2n = 4x = 8 = 6sm + 2st; Figure 3G,H and Figure S4; Table 2). This type of karyotype formula was recovered for C. aculeata and C. setosa (subclade IVb; Figure S4; Table 2). In subclade IVa, all of the species had the karyotype formula of 2n = 2x = 8 = 2sm + 6st (Figure 5 and Figure 3A; Table 2; Figure S4). Species with the karyotype formula 2n = 2x = 8 = 4sm + 4st were present in subclades IVb (C. kotschyana, C. aspera) and IVc (C. alpestris; Figure 5 and Figure S4; Table 2). Five species with a chromosome base number of x = 4 had karyotypes with metacentric, submetacentric, and subtelocentric chromosomes. The karyotype formula 2n = 2x = 2m + 2sm + 4st was present in two species from subclade IVc (C. oporinoides and C. pyrenaica) and three species from the Lagoseris evolutionary lineage (C. palaestina, C. pulchra, and C. praemorsa; Figure 5 and Figure S4; Table 2). Two species with a chromosome base number x = 3 belonged to two different subclades (Figure 5) and had two different karyotype formulas (2n = 2x = 6 = 2m + 4sm for C. zacintha; Figure 3C; Table 2; and 12n = 2x = 6 = 2sm + 4st for C. capillaris; Figure 3J; Table 2).
3.4. Genome Size
The genome sizes (1C values) of the 33 Crepis species and for L. communis were measured (Table 2). The genome sizes varied nearly seven-fold among the diploid species, ranging from 1.03 pg/1C DNA (C. zacintha) to 7.46 pg/1C DNA (C. lacera). The genome sizes of the species of the Lagoseris evolutionary lineage were variable with nearly a six-fold difference (1.22 pg/1C DNA to 7.05 pg/1C DNA). In Crepis s.s., larger genome sizes were found in two species from clade I (mean 1C value = 4.78 pg), whereas clade II species had smaller genome sizes (mean 1C value = 2.44 pg). In clade III, two diploid species C. tectorum (3.06 pg/1C DNA) and C. nicaeensis (3.17 pg/1C DNA) had small genomes, whereas the octoploid C. biennis had a larger genome (10.45 pg/1C DNA). The Cx-value (the C-value of monoploid genome “x”) of C. biennis was rather small (1Cx = 2.61 pg/1C DNA; Figure 5 and Figure S7) compared to the diploid species of this group. Three different groups of related species that differed in the patterns of their genome sizes distribution were identified in clade IV. The subclade IVa consisted of diploid species with larger genome sizes (mean 1C value = 6.35 pg) and, among them, C. lacera had the largest estimated genome size (1C value = 7.46 pg). The genome sizes of the species from clade IVc were on average 2.52 pg/1C DNA. The largest variation in genome sizes was observed among the species in subclade IVb in which the genome size of C. sibirica (1C value = 6.98 pg) was nearly seven times larger than that of C. zacintha (1C value = 1.03 pg; Table 2; Figure 5 and Figure S7). C. vesicaria contained both diploid and tetraploid accessions (2n = 8 and 2n = 16). The genome sizes that were estimated for the diploid and tetraploid accessions (2.43 pg/1C and 2.78 pg/1C, respectively).
The DNA amounts were mapped onto the cpDNA ML phylogenetic tree using the maximum likelihood method (Figure 5 and Figure S7). A genome size of 3.2 pg/1C DNA was inferred as the ancestral state for the Lagoseris lineage. Both decreases and increases in genome sizes were inferred to have accompanied speciation and diversification of this clade (Figure 5 and Figure S7). The ancestral genome size for the entire Crepis s.s. lineage was reconstructed as 3.70 pg/1C. For clade I, 4.8 pg/1C was reconstructed as the ancestral state, whereas 2.50 pg/1C was recovered as the ancestral genome size for clade II. For the entire clade IV, 3.2 pg/1C was reconstructed as the ancestral genome size. For subclade IVa, the ancestral genome size was reconstructed to be 5.5 pg/1C, whereas for subclades IVb and IVc, 2.7 pg/1C and 2.9 pg/1C, respectively, were recovered. An increase in genome size was inferred for the species of clade I and the octoploid C. biennis (clade III). In contrast, a reduction in genome size accompanied the evolution of many species of clade II and subclades IVb and IVc (e.g., C. aurea and C. zacintha). The evolution of a common ancestor of subclade IVa was accompanied by an increase in the genome size, with further increases or decreases in the genome sizes inferred during the diversification and speciation of this clade.
The inferences of genome size evolution were additionally performed using nrITS tree based on an extended data set containing both newly obtained genome size data and previously reported genome size estimations (Figure S8; Table S2). The extended data set included the species from the American agamic complex, which had relatively large genome sizes (Table S3). The observed values of the reconstructed ancestral states were slightly larger in comparison to the results of the analyses of the newly obtained data only. The inferred events of increases and reductions in genome size were very similar in both data sets.
4. Discussion
Resolving the phylogenetic relationships for Crepis provided the evolutionary framework for elucidating the evolution of the chromosome numbers and karyotype structure. Phylogeny based on the biparentally inherited nrITS of mostly diploid species of Crepis was found to be largely congruent with previously published results (Figure S1; [22,23]). In the present study, a new clade comprising species of the North American Crepis agamic complex was identified. These species have not been included in previous analyses. The phylogenetic relationships inferred in this study were inconsistent with the sections defined by Babcock [20], in agreement with the earlier phylogenetic analyses [22]. Each clade and subclade contained species previously classified in different Babcock sections (Figure S1). A phylogenetic analysis, based on the chloroplast markers, recovered mostly the same main groups of species as the nrITS-based analyses. However, the relationships among these clades were found to be incongruent between the nuclear and plastid markers. Such nucleo-cytoplasmic discordance has been well documented in previous studies of higher plants [49]. This incongruence could be explained by the convergent evolution of shared chloroplast sequences, incomplete lineage sorting of the ancestral polymorphisms, or introgressive hybridization [50,51]. The Crepis species analysed here were mostly diploids, but because Crepis species are primarily cross-pollinated and many of them hybridise extensively [20], introgression could have accompanied the speciation of individual species, as also suggested for other plant groups [52,53]. The incongruence could be also explained by technical reasons. Several nodes in nrITS phylogram in clade 4 were weakly supported and the analysed species represent only a quarter of the Crepis genome, which may influence the results. The incongruence between the nrITS and cpDNA trees concerning the position of C. biennis could suggest its allopolyploid origin. C. biennis is an octoploid species [47] whose origin and parental species are not yet known. The present analysis suggests a putative ancestral species for the two subgenomes of this polyploid taxon, species that are similar to the extant C. nicaeensis/C. setosa and another species related to C. tectorum (Figure 1). Additional molecular phylogenetic and cytogenetic studies are necessary to elucidate the origin of C. biennis.
This study reports the genome sizes for 33 species of Crepis s.l., eight of them for the first time. The genome sizes that were reported in the current study are largely in agreement with previous reports, except for two species: C. vesicaria and C. pannonica (Table 2; [12,54,55]). This discrepancy is probably the result of methodological factors as the previous data were obtained using Feulgen densitometry or flow cytometry but with different standards. In the case of the diploid C. vesicaria, the discrepancy (2.43 pg/1C in this study versus 4.18 reported by Enke et al. [12]) could result from the presence of the diploid and polyploid cytotypes within this species [20]. A strong correlation between genome size and life form has been suggested for Crepis in previous studies [12]. The current results, however, did not show an obvious correlation between these two characters. Both annual and perennial Crepis species groups showed high levels of variation of their genome sizes. Both one of the smallest (C. leontodontoides 1.06 pg/1C) and the largest genomes (C. lacera; 7.46 pg/1C) were found in the perennial diploid species. The correlation between genome size and plant life form has been extensively discussed in the literature. The annual life form, especially in weedy species, was inferred to be associated with a smaller genome size compared to its related perennials [56,57,58]. Such a correlation, however, must be interpreted with caution because other ecological and evolutionary factors might also influence genome size evolution [59]. The chromosome numbers of Crepis species analysed in the current study were in accordance with earlier reports [20,60]. Most of the analysed species were diploids, with only a few polyploids [20]. The tetraploids of C. vesicaria shared the same karyotype structure with its diploid accession, which suggests that they were of autopolyploid origin
Analyses of chromosome number and genome size evolution in a phylogenetic framework revealed that there was no general trend in their evolution among angiosperms [61,62,63]. Both chromosome numbers and genome sizes could change very rapidly during the evolution and diversification of plant genomes [64,65,66]. A reduction in genome size was earlier suggested to accompany the evolution of the genus Crepis [12,67], with increases in genome sizes mainly correlated with polyploidisation [20]. However, the present data revealed a bidirectional evolution of genome size in both the Lagoseris and Crepis s.s. evolutionary lineages. The current study revealed that several diploid species have a genome size that is much larger than the ancestral reconstructed states of the lineages they belong to, e.g., C. pannonica or C. pulchra. This suggested a bidirectional evolution of genome size in at least some of the Crepis lineages. The proliferation of repetitive DNA sequences is considered to be the major cause of the increase in genome size next to polyploidy [7,24,68]. The mechanisms of the reduction of genome sizes and DNA removal are less understood, with an unequal homologous recombination or illegitimate recombination proposed to be most important [69,70]. The information about the genome structure in Crepis is still very limited and only further analyses, especially of repetitive DNA fraction, will allow better insight into the mechanisms of the evolution of genome size in this genus.
The ancestral states of the base chromosome number in a common ancestor of Lagoseris and Crepis s.s. was inferred to be x = 6, and descending disploidy was inferred as a main trend in the chromosome number evolution in Crepis (Figure 4). The obtained results are partly in agreement with the hypothesis of Babcock [20], who suggested that a decrease in the chromosome base number is the main trend in Crepis evolution. However, the derived base chromosome numbers (x = 5, 4, and 3) were found to have evolved independently several times in the evolutionary history of the genus. The chromosome base number of x = 5 has evolved at least three times during Crepis evolution. Dysploid change from x = 5 to x = 4 has occurred twice in Lagoseris and eight times in Crepis s.s. The base chromosome x = 3 has evolved twice in Crepis s.s. Nearly half of the descending dysploidy events were recovered for the same branches for which changes in genome sizes were inferred (Figure 5). Some descending dysploidy events were accompanied by a reduction in genome sizes whereas others were accompanied by genome size increases and no common trend was observed. Similar patterns have been reported for Artemisia (Asteraceae; [64]) and other plant genera (e.g., Genlisea, Lentibulariaceae; [65]).
The chromosome base number was inferred to have increased only twice during the evolution of the analysed species. An allopolyploidisation or a duplication of the chromosome number followed by dysploidy is likely to have given rise to the base chromosome of x = 11 present in the American agamic complex. The evolution of this lineage was accompanied by an increase in genome size (Figure S8), supporting the hypothesis of the polyploidy origin of this new base chromosome number of x = 11. In contrast, the increase in base chromosome number in the evolutionary lineage of Lapsana (from x = 6 to x = 7) was accompanied by the genome size downsizing. The chromEvol analyses inferred that the changes in chromosome number in the Lapsana lineage resulted from demiduplication. For demiduplication, a genome increase instead of decrease would be expected. Unfortunately, the direct ancestor of Lapsana and its genome size are unknown and thus this hypothesis cannot be tested. More detailed comparative analyses of the karyotype structure of Lapsana and related taxa are necessary to gain insight into the origin of base chromosome number of x = 7 in the Lagoseris lineage. Changes in the chromosome base numbers as well as in genome sizes in Crepis were most prevalent at the tips of the tree rather than early in the evolution of the genus. Similar trends have been reported for Trifolium, Hypochaeris, or Braschyscome [71,72,73].
Chromosomal rearrangements often accompany plant diversification and speciation [74,75,76]. Chromosome rearrangements generally have a low level of homoplasy. Therefore, it was hypothesised that species with the same chromosome number that evolved independently should differ in their chromosome structure [1]. Molecular cytogenetic methods and the availability of many chromosomal markers enables the comparative analysis of the karyotype in model or cultivated taxa (e.g., Brassicaceae and Brachypodium [77,78,79]. In wild species groups it is more challenging, but even comparative analyses of the morphology of chromosomes are indicative of occurrences of chromosomal rearrangements. However, the shape and size of the chromosomes can also be altered by a sequence amplification/loss [1,7,8,9]. A comparative analysis of the Crepis karyotype formulas revealed that species with the same chromosome base number belonging to different evolutionary lineages differed in their karyotype morphologies (Figure 5). Moreover, even the species in the same subclades (e.g., subclade IVc) possessing the same chromosome base number differed in karyotype structure (Figure 5). These results supported the hypothesis that the derived base chromosome numbers (x = 5, 4, 3) might have evolved independently multiple times in Crepis. The changes in karyotype structure might lead to an increase in its asymmetry [38]. An increase in karyotype asymmetry was previously suggested as the main pattern of karyotype evolution in the genus Crepis [80]. The results of the present study did not support this hypothesis. An increase in karyotype asymmetry accompanied the speciation of only a few species in the different evolutionary lineages (Figure 5). The increases in karyotype asymmetry accompanied both decreases and an increase in the chromosome base numbers (Figure 5), as well as increases or decreases in the genome sizes. Some groups of closely related Crepis species had the same chromosome number and a similar karyotype structure (e.g., subclade IVa), which suggests that different mechanisms, e.g., an amplification of repetitive sequences, might have accompanied the diversification of these species.
5. Conclusions
Analyses of karyotype structure and genome sizes in a phylogenetic context enabled inferences about the ancestral base chromosome numbers and genome sizes in the Lagoseris and Crepis s.s. evolutionary lineages. A few events of base chromosome number changes, mostly descending dysploidy, explained the chromosome number distribution in the extant Crepis s.l. species best. Most of these changes accompanied the evolution of individual species or a small group of closely related, derived species. Analyses of the genome size evolution revealed both increases and decreases during the evolution of the genus. The changes in genome sizes occurred mainly at the tips rather than at the internal nodes of the tree. The present work contributes to a better understanding of the evolution of the genomes of Crepis and lays the foundation for more detailed comparative analyses of its karyotype structure and evolution, including repetitive sequences.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/genes12091436/s1, Figure S1: Maximum likelihood phylogenetic tree based on nrITS sequences incorporating of taxa analysed in the present study and available in GenBank accessions of other species from Crepis s.l. and related taxa. All the species analysed in this study are indicated in bold. Bootstrap support values are indicated at each node, Figure S2: Metaphase plates of Crepis species used to prepare the karyograms in Figure 2 (A) Lapsana communis, (B) C. magellensis, (C) C. jacquinni, (D) C. paludosa, (E) C. succisifolia, (F) C. lyrata, (G) C. nicaeensis. Scale bar = 5 µm, Figure S3: Metaphase plates of Crepis species used to prepare the karyograms in Figure 3: (A) C. pannonica, (B) C. foetida subsp. rhoaedifolia, (C) C. zacintha, (D) C. syriaca, (E) C. kotschyana, (F) C. vesicaria 2, (G) C. vesicaria 1, (H) C. leontodontoides, (I) C. oporinoides, (J) C. capillaris. Scale bar = 5 µm, Figure S4: Metaphase chromosomes of analysed Crepis species: (A) C. sancta, (B) C. palaestina, (C) C. pulchra, (D) C. praemorsa, (E) C. mollis, (F) C. pygmae, (G) C. tectorum, (H) C. nigrescens, (I) C. dioscoridis, (J) C. conyzifolia dshimilensis, (K) C. lacera, (L) C. conyzifolia, (M) C. foetida, (N) C. rubra, (O) C. setosa, (P) C. sibirica, (Q) C. alpina, (R) C. aspera, (S) C. aculeata, (T) C. vesicaria 3, (U) C. taraxacifolia, (V) C. polymorpha, (W) C. albida, (X) C. aurea, (Y) C. pyrenaica, (Z) C. alpestris. Descriptions below each pair of chromosomes indicate the type of chromosome: m—metacentric; sm—submetacentric; and st—subtelocentric. The metaphase plates used to prepare karyograms are presented in Figure S9. Scale bar = 5 µm, Figure S5: Metaphase plate of Crepis occidentalis (A) and C. biennis (B), Figure S6: Ancestral character state reconstruction of the asymmetry indexes of karyotypes of analysed species of Crepis s.l. The asymmetry indexes have been mapped on the ML tree of concatenated cpDNA sequences using maximum likelihood method as implemented in phytools. The tree was rooted with Picris hieracioides, Lactuca serriola, and Sonchus oleraceus, Figure S7: Ancestral character state reconstruction of the genome size (1C) of species of Crepis s.l. The 1C DNA amounts have been mapped on the ML tree of cpDNA sequences using maximum likelihood reconstruction as implemented in phytools. Numbers next to the nodes indicate reconstructed ancestral 1C-values (in pg). The genome size of each species is presented in Table 2. The tree was rooted with Picris hieracioides, Lactuca serriola, and Sonchus oleraceus, Figure S8: Ancestral character state reconstruction of genome size (1C-value) of Crepis s.l. taxa analysed in the present study and available in literature. The analysis was conducted using maximum likelihood reconstruction as implemented in phytools, Figure S9: Metaphase plates of Crepis species used to prepare the karyograms in Figure S4: (A) C. sancta, (B) C. palaestina, (C) C. pulchra, (D) C. praemorsa, (E) C. mollis, (F) C. pygmae, (G) C. tectorum, (H) C. nigrescens, (I) C. dioscoridis, (J) C. conyzifolia dshimilensis, (K) C. lacera, (L) C. conyzifolia, (M) C. foetida, (N) C. rubra, (O) C. setosa, (P) C. sibirica, (Q) C. alpina, (R) C. aspera, (S) C. aculeata, (T) C. vesicaria 3, (U) C. taraxacifolia, (V) C. polymorpha, (W) C. albida, (X) C. aurea, (Y) C. pyrenaica, (Z) C. alpestris. Scale bar = 5 µm, Table S1: Sequences of primers used for PCR amplification and sequencing, Table S2: Species name and GenBank accessions numbers of the selected Crepis sequences from Enke et al. [22] used in this study, Table S3: Genome size of Crepis species analysed in present study and data retrieved from literature, Table S4: The ΔAIC scores and Akaike weights of each model tested in ChromEvol 2.0 software for nrITS and cpDNA data sets, Table S5: Length of analysed plastid DNA and nrITS (nuclear ITS: ITS1-5.8S rDNA-ITS2) data sets [81,82].
Author Contributions
B.K. conceived and designed the study; M.S., M.B., M.R.-J. and B.K. performed the laboratory work and subsequent analyses; T.N. and L.P. collected plant material; B.K., H.W.-S. and M.S. writing—original draft preparation; B.K., H.W.-S. and M.S. writing—original draft preparation. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre, Poland (Project No. 2017/27/B/NZ8/01478).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All data generated or analysed during this study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schubert, I.; Vu, G.T.H. Genome Stability and Evolution: Attempting a Holistic View. Trends Plant Sci. 2016, 21, 749–757. [Google Scholar] [CrossRef]
- Lysák, M.A.; Schubert, I. Mechanisms of Chromosome Rearrangements. In Plant Genome Diversity Volume 2: Physical Structure, Behaviour and Evolution of Plant Genomes; Greilhuber, J., Dolezel, J., Wendel, J.F., Eds.; Springer Vienna: Vienna, Austria, 2013; pp. 137–147. [Google Scholar] [CrossRef]
- Coghlan, A.; Eichler, E.E.; Oliver, S.G.; Paterson, A.H.; Stein, L. Chromosome evolution in eukaryotes: A multi-kingdom perspective. Trends Genet. 2005, 21, 673–682. [Google Scholar] [CrossRef]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Anderson, L.K.; Covey, P.A.; Larsen, L.R.; Bedinger, P.; Stack, S.M. Structural differences in chromosomes distinguish species in the tomato clade. Cytogenet. Genome Res. 2010, 129, 24–34. [Google Scholar] [CrossRef]
- Baeza, C.; Finot, V.L.; Ruiz, E. Comparative karyotype analysis of populations in the Alstroemeria presliana Herbert (Alstroemeriaceae) complex in Chile. Genet. Mol. Biol. 2015, 38, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Ambrožová, K.; Mandáková, T.; Bureš, P.; Neumann, P.; Leitch, I.J.; Koblížková, A.; Macas, J.; Lysak, M.A. Diverse retrotransposon families and an AT-rich satellite DNA revealed in giant genomes of Fritillaria lilies. Ann. Bot. 2011, 107, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Weiss-Schneeweiss, H.; Schneeweiss, G.M. Karyotype Diversity and Evolutionary Trends in Angiosperms. In Plant Genome Diversity Volume 2: Physical Structure, Behaviour and Evolution of Plant Genomes; Greilhuber, J., Dolezel, J., Wendel, J.F., Eds.; Springer Vienna: Vienna, Austria, 2013; pp. 209–230. [Google Scholar] [CrossRef]
- Li, S.F.; Su, T.; Cheng, G.Q.; Wang, B.X.; Li, X.; Deng, C.L.; Gao, W.J. Chromosome Evolution in Connection with Repetitive Sequences and Epigenetics in Plants. Genes 2017, 8, 290. [Google Scholar] [CrossRef]
- Funk, V.A.; Susanna, A.; Stuessy, T.F.; Bayer, R.J. Systematics, Evolution, and Biogeography of Compositae; Smithsonian Institution: Washington, DC, USA, 2009. [Google Scholar]
- Babcock, E.B.; Jenkins, J.A. Chromosomes and Phylogeny in Crepis, III: The Relationships of one Hundred and Thirteen Species; University of California Press: Berkeley, CA, USA, 1943. [Google Scholar]
- Enke, N.; Fuchs, J.; Gemeinholzer, B. Shrinking genomes? Evidence from genome size variation in Crepis (Compositae). Plant Biol. 2011, 13, 185–193. [Google Scholar] [CrossRef]
- Stebbins, G.L. Chromosomal Evolution in Higher Plants; Edward Arnold: London, UK, 1971. [Google Scholar]
- Smocovitis, V.B. The “Plant Drosophila”: E.B. Babcock, the genus “Crepis”, and the evolution of a genetics research program at Berkeley, 1915–1947. Hist. Stud. Nat. Sci. 2009, 39, 300–355. [Google Scholar] [CrossRef]
- Hollingshead, L.; Babcock, E. Chromosomes and Phylogeny in Crepis; University of California Press: Berkeley, CA, USA, 1930; Volume 6, pp. 1–53. [Google Scholar]
- Babcock, E.B.; Cameron, D.R. Chromosomes and Phylogeny in Crepis. II. The Relationships of one Hundred Eight Species; University of California Press: Berkeley, CA, USA, 1934; pp. 287–324. [Google Scholar]
- Da Silva, C.R.M.; Gonzalez-Elizondo, M.S.; Vanzela, A.L.L. Reduction of chromosome number in Eleocharis subarticulata (Cyperaceae) by multiple translocations. Bot. J. Linn. Soc. 2005, 149, 457–464. [Google Scholar] [CrossRef]
- Wan, T.; Zhang, X.; Gregan, J.; Zhang, Y.; Guo, P.; Guo, Y. A dynamic evolution of chromosome in subgenus Potamogeton revealed by physical mapping of rDNA loci detection. Plant Syst. Evol. 2012, 298, 1195–1210. [Google Scholar] [CrossRef]
- Briggs, D.; Walters, S.M. Plant Variation and Evolution, 4th ed.; Cambridge University Press: Cambridge, UK, 2016. [Google Scholar] [CrossRef]
- Babcock, E.B. The Genus Crepis I. The Taxonomy, Phylogeny, Distribution and Evolution of Crepis. The Genus Crepis II. Systematic treatment.; University of California Press: Berkeley, CA, USA, 1947. [Google Scholar]
- Levitsky, G.A. The karyotype in systematics. Bull. Appl. Bot. Genet. Plant Breed. 1931, 27, 220–240. [Google Scholar]
- Enke, N.; Gemeinholzer, B. Babcock revisited: New insights into generic delimitation and character evolution in Crepis L. (Compositae: Cichorieae) from ITS and matK sequence data. Taxon 2008, 57, 756–768. [Google Scholar] [CrossRef]
- Enke, N. Contributions towards a revised infrageneric classification of Crepis (Cichorieae, Compositae). Willdenowia 2010, 39, 229–245. [Google Scholar] [CrossRef]
- Emadzade, K.; Jang, T.-S.; Macas, J.; Kovařík, A.; Novák, P.; Parker, J.; Weiss-Schneeweiss, H. Differential amplification of satellite PaB6 in chromosomally hypervariable Prospero autumnale complex (Hyacinthaceae). Ann. Bot. 2014, 114, 1597–1608. [Google Scholar] [CrossRef]
- Venora, G.; Blangiforti, S.; Frediani, M.; Maggini, F.; Gelati, M.T.; Castiglione, M.R.; Cremonini, R. Nuclear DNA contents, rDNAs, chromatin organization, and karyotype evolution inVicia sect, faba. Protoplasma 2000, 213, 118–125. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar] [CrossRef]
- Shaw, J.; Shafer, H.L.; Leonard, O.R.; Kovach, M.J.; Schorr, M.; Morris, A.B. Chloroplast DNA sequence utility for the lowest phylogenetic and phylogeographic inferences in angiosperms: The tortoise and the hare IV. Am. J. Bot. 2014, 101, 1987–2004. [Google Scholar] [CrossRef]
- Shaw, J.; Lickey, E.B.; Schilling, E.E.; Small, R.L. Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: The tortoise and the hare III. Am. J. Bot. 2007, 94, 275–288. [Google Scholar] [CrossRef]
- Blöch, C.; Weiss-Schneeweiss, H.; Schneeweiss, G.M.; Barfuss, M.H.J.; Rebernig, C.A.; Villaseñor, J.L.; Stuessy, T.F. Molecular phylogenetic analyses of nuclear and plastid DNA sequences support dysploid and polyploid chromosome number changes and reticulate evolution in the diversification of Melampodium (Millerieae, Asteraceae). Mol. Phylogenet. Evol. 2009, 53, 220–233. [Google Scholar] [CrossRef]
- Löytynoja, A.; Goldman, N. webPRANK: A phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinform. 2010, 11, 579. [Google Scholar] [CrossRef]
- Collingridge, P.W.; Kelly, S. MergeAlign: Improving multiple sequence alignment performance by dynamic reconstruction of consensus multiple sequence alignments. BMC Bioinform. 2012, 13, 117. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2017, 35, 518–522. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.2, A Graphical Viewer of Phylogenetic Trees. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 1 September 2014).
- Dydak, M.; Kolano, B.; Nowak, T.; Siwinska, D.; Maluszynska, J. Cytogenetic studies of three European species of Centaurea L. (Asteraceae). Hereditas 2009, 146, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Levan, A.; Fredga, K.; Sandberg, A.A. Nomenclature for centromeric position on chromosomes. Hereditas 1964, 52, 201–220. [Google Scholar] [CrossRef]
- Paszko, B. A critical review and a new proposal of karyotype asymmetry indices. Plant Syst. Evol. 2006, 258, 39–48. [Google Scholar] [CrossRef]
- Catalán, P.; Müller, J.; Hasterok, R.; Jenkins, G.; Mur, L.A.J.; Langdon, T.; Betekhtin, A.; Siwinska, D.; Pimentel, M.; López-Alvarez, D. Evolution and taxonomic split of the model grass Brachypodium distachyon. Ann. Bot. 2012, 109, 385–405. [Google Scholar] [CrossRef] [PubMed]
- Doležel, J.; Sgorbati, S.; Lucretti, S. Comparison of three DNA fluorochromes for flow cytometric estimation of nuclear DNA content in plants. Physiol. Plant. 1992, 85, 625–631. [Google Scholar] [CrossRef]
- Temsch, E.M.; Greilhuber, J.; Krisai, R. Genome size in liverworts. Preslia 2010, 82, 63–80. [Google Scholar]
- Lysak, M.A.; Dolezel, J. Estimation of nuclear DNA content in Sesleria (Poaceae). Caryologia 1998, 51, 123–132. [Google Scholar] [CrossRef]
- Doležel, J.; Greilhuber, J.; Lucretti, S.; Meister, A.; Lysák, M.A.; Nardi, L.; Obermayer, R. Plant genome size estimation by flow cytometry: Inter-laboratory comparison. Ann. Bot. 1998, 82, 17–26. [Google Scholar] [CrossRef]
- Greilhuber, J.; Dolezel, J.; Lysák, M.A.; Bennett, M.D. The origin, evolution and proposed stabilization of the terms ‘genome size’ and ‘C-value’ to describe nuclear DNA contents. Ann. Bot. 2005, 95, 255–260. [Google Scholar] [CrossRef]
- Glick, L.; Mayrose, I. ChromEvol: Assessing the pattern of chromosome number evolution and the inference of polyploidy along a phylogeny. Mol. Biol. Evol. 2014, 31, 1914–1922. [Google Scholar] [CrossRef]
- Babcock, E.; Stebbins, G. The American Species of Crepis Their Interrelationships and Distribution as Affected by Polyploidy and Apomixis; Carnegie Institution of Washington: Washington, DC, USA, 1938. [Google Scholar] [CrossRef]
- Babcock, E.B.; Swezy, O. The Chromosomes of Crepis biennis L., and Crepis ciliata C. Koch. Cytologia 1935, 6, 256–265. [Google Scholar] [CrossRef]
- Revell, L.J. Phytools: An R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 2012, 3, 217–223. [Google Scholar] [CrossRef]
- Bonnet, T.; Leblois, R.; Rousset, F.; Crochet, P.-A. A reassessment of explanations for discordant introgressions of mitochondrial and nuclear genomes. Evolution 2017, 71, 2140–2158. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Acosta, C.M.; Premoli, A.C. Evidence of chloroplast capture in South American Nothofagus (subgenus Nothofagus, Nothofagaceae). Mol. Phylogenetics Evol. 2010, 54, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Eaton, D.A.R.; Ree, R.H. Inferring Phylogeny and Introgression using RADseq Data: An Example from Flowering Plants (Pedicularis: Orobanchaceae). Syst. Biol. 2013, 62, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Gaudeul, M.; Gardner, M.F.; Thomas, P.; Ennos, R.A.; Hollingsworth, P.M. Evolutionary dynamics of emblematic Araucaria species (Araucariaceae) in New Caledonia: Nuclear and chloroplast markers suggest recent diversification, introgression, and a tight link between genetics and geography within species. BMC Evol. Biol. 2014, 14, 171. [Google Scholar] [CrossRef]
- Bennett, M.D.; Smith, J.B.; Riley, R. Nuclear DNA amounts in angiosperms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1976, 274, 227–274. [Google Scholar] [CrossRef] [PubMed]
- Smarda, P.; Knapek, O.; Brezinova, A.; Horova, L.; Grulich, V.; Danihelka, J.; Vesely, P.; Smerda, J.; Rotreklova, O.; Bures, P. Genome sizes and genomic guanine plus cytosine (GC) contents of the Czech vascular flora with new estimates for 1700 species. Preslia 2019, 91, 117–142. [Google Scholar] [CrossRef]
- Bennett, M.D.; Riley, R. Nuclear DNA content and minimum generation time in herbaceous plants. Proc. Biol. Sci. 1972, 181, 109–135. [Google Scholar] [CrossRef]
- Garnatje, T.; Vallès, J.; Garcia, S.; Hidalgo, O.; Sanz, M.; Canela, M.Á.; Siljak-Yakovlev, S. Genome size in Echinops L. and related genera (Asteraceae, Cardueae): Karyological, ecological and phylogenetic implications. Biol. Cell 2004, 96, 117–124. [Google Scholar] [CrossRef]
- Weiss-Schneeweiss, H.; Greilhuber, J.; Schneeweiss, G.M. Genome size evolution in holoparasitic Orobanche (Orobanchaceae) and related genera. Am. J. Bot. 2006, 93, 148–156. [Google Scholar] [CrossRef]
- Andrés-Sánchez, S.; Temsch, E.M.; Rico, E.; Montserrat Martínez-Ortega, M. Genome size in Filago L. (Asteraceae, Gnaphalieae) and related genera: Phylogenetic, evolutionary and ecological implications. Plant Syst. Evol. 2013, 299, 331–345. [Google Scholar] [CrossRef]
- Sears, C.J.; Whitton, J. A reexamination of the North American Crepis agamic complex and comparison with the findings of Babcock and Stebbins’ classic biosystematic monograph. Am. J. Bot. 2016, 103, 1289–1299. [Google Scholar] [CrossRef]
- Winterfeld, G.; Paule, J.; Hoffmann, M.H.; Ley, A.; Röser, M. Antagonistic effects of whole-genome duplications and dysploidy on genome sizes in the pantropical monocot family Marantaceae: Consequences in the light of a new molecular phylogeny. Curr. Plant Biol. 2020, 24, 100181. [Google Scholar] [CrossRef]
- Vitales, D.; Álvarez, I.; Garcia, S.; Hidalgo, O.; Nieto Feliner, G.; Pellicer, J.; Vallès, J.; Garnatje, T. Genome size variation at constant chromosome number is not correlated with repetitive DNA dynamism in Anacyclus (Asteraceae). Ann. Bot. 2019, 125, 611–623. [Google Scholar] [CrossRef]
- Pellicer, J.; Kelly, L.J.; Leitch, I.J.; Zomlefer, W.B.; Fay, M.F. A universe of dwarfs and giants: Genome size and chromosome evolution in the monocot family Melanthiaceae. N. Phytol. 2014, 201, 1484–1497. [Google Scholar] [CrossRef]
- Mas de Xaxars, G.; Garnatje, T.; Pellicer, J.; Siljak-Yakovlev, S.; Vallès, J.; Garcia, S. Impact of dysploidy and polyploidy on the diversification of high mountain Artemisia (Asteraceae) and allies. Alp. Bot. 2016, 126, 35–48. [Google Scholar] [CrossRef]
- Fleischmann, A.; Michael, T.P.; Rivadavia, F.; Sousa, A.; Wang, W.; Temsch, E.M.; Greilhuber, J.; Müller, K.F.; Heubl, G. Evolution of genome size and chromosome number in the carnivorous plant genus Genlisea (Lentibulariaceae), with a new estimate of the minimum genome size in angiosperms. Ann. Bot. 2014, 114, 1651–1663. [Google Scholar] [CrossRef]
- Winterfeld, G.; Ley, A.; Hoffmann, M.H.; Paule, J.; Röser, M. Dysploidy and polyploidy trigger strong variation of chromosome numbers in the prayer-plant family (Marantaceae). Plant Syst. Evol. 2020, 306, 36. [Google Scholar] [CrossRef]
- Enke, N.; Kunze, R.; Pustahija, F.; Glöckner, G.; Zimmermann, J.; Oberländer, J.; Kamari, G.; Siljak-Yakovlev, S. Genome size shifts: Karyotype evolution in Crepis section Neglectoides (Asteraceae). Plant Biol. 2015, 17, 775–786. [Google Scholar] [CrossRef]
- Vitte, C.; Panaud, O. LTR retrotransposons and flowering plant genome size: Emergence of the increase/decrease model. Cytogenet. Genome Res. 2005, 110, 91–107. [Google Scholar] [CrossRef] [PubMed]
- Bennetzen, J.L. Modes, rates and mechanisms of local genomic change in flowering plants: Tempest in a pea plot. In Plant Species-Level Systematics: New Perspectives on Pattern & Process; Bakker, F.T., Chatrou, L.W., Gravendeel, B., Pelser, P.B., Eds.; A.R.G. Gantner Verlag: Ruggell, Liechtenstein, 2005; p. 348. [Google Scholar]
- Bennetzen, J.L.; Wang, H. The contributions of transposable elements to the structure, function, and evolution of plant genomes. Annu. Rev. Plant Biol. 2014, 65, 505–530. [Google Scholar] [CrossRef]
- Ellison, N.W.; Liston, A.; Steiner, J.J.; Williams, W.M.; Taylor, N.L. Molecular phylogenetics of the clover genus (Trifolium—Leguminosae). Mol. Phylogenetics Evol. 2006, 39, 688–705. [Google Scholar] [CrossRef] [PubMed]
- Cerbah, M.; Coulaud, J.; Brown, S.; Siljak-yakovlev, S. Evolutionary DNA variation in the genus Hypochaeris. Heredity 1999, 82, 261–266. [Google Scholar] [CrossRef]
- Watanabe, K.; Yahara, T.; Denda, T.; Kosuge, K. Chromosomal Evolution in the Genus Brachyscome (Asteraceae, Astereae): Statistical Tests Regarding Correlation Between Changes in Karyotype and Habit Using Phylogenetic Information. J. Plant. Res. 1999, 112, 145–161. [Google Scholar] [CrossRef]
- Shan, F.; Yan, G.; Plummer, J.A. Karyotype evolution in the genus Boronia (Rutaceae). Bot. J. Linn. Soc. 2003, 142, 309–320. [Google Scholar] [CrossRef]
- Mandáková, T.; Lysak, M.A. Chromosomal phylogeny and karyotype evolution in x = 7 crucifer species (Brassicaceae). Plant Cell 2008, 20, 2559–2570. [Google Scholar] [CrossRef] [PubMed]
- Baltisberger, M.; Hörandl, E. Karyotype evolution supports the molecular phylogeny in the genus Ranunculus (Ranunculaceae). Perspect. Plant Ecol. Evol. Syst. 2016, 18, 1–14. [Google Scholar] [CrossRef][Green Version]
- Lysak, M.A.; Koch, M.A. Phylogeny, Genome, and Karyotype Evolution of Crucifers (Brassicaceae). In Genetics and Genomics of the Brassicaceae. Plant Genetics and Genomics: Crops and Models; Schmidt, R., Bancroft, I., Eds.; Springer: New York, NY, USA, 2011. [Google Scholar]
- Lysak, M.A.; Berr, A.; Pecinka, A.; Schmidt, R.; McBreen, K.; Schubert, I. Mechanisms of chromosome number reduction in Arabidopsis thaliana and related Brassicaceae species. Proc. Natl. Acad. Sci. USA 2006, 103, 5224–5229. [Google Scholar] [CrossRef]
- Lusinska, J.; Betekhtin, A.; Lopez-Alvarez, D.; Catalan, P.; Jenkins, G.; Wolny, E.; Hasterok, R. Comparatively Barcoded Chromosomes of Brachypodium Perennials Tell the Story of Their Karyotype Structure and Evolution. Int. J. Mol. Sci. 2019, 20, 5557. [Google Scholar] [CrossRef]
- Stebbins, G.L. Chromosomal Variation and Evolution. Science 1966, 152, 1463–1469. [Google Scholar] [CrossRef]
- Wallace, H.; Sparkes, C.A.; Maden, M. Nuclear DNA content of three Crepis species. Heredity 1972, 29, 367–373. [Google Scholar] [CrossRef][Green Version]
- Dimitrova, D.; Greilhuber, J. Karyotype and DNA-content evolution in ten species of Crepis (Asteraceae) distributed in Bulgaria. Bot. J. Linn. Soc. 2000, 132, 281–297. [Google Scholar] [CrossRef][Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).