
Resistance of the Wheat Cultivar ‘Renan’ to Septoria Leaf Blotch Explained by a Combination of Strain Specific and Strain Non-Specific QTL Mapped on an Ultra-Dense Genetic Map


 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant and Fungal Materials
2.2. Experimental Setup and Procedure for Pathology Assays
2.2.1. Data Sets
2.2.2. Culture Conditions
2.2.3. Inoculum Preparation
2.2.4. Inoculation
2.3. Evaluation of Phenotypic Traits
2.3.1. Visual Evaluation of Symptoms
2.3.2. Pycnidia Counting by Image Analysis
2.3.3. Quantification of Sporulation
2.4. Statistical Analysis of Phenotypic Data
2.5. Genotyping RxCS
2.5.1. Axiom 410K
2.5.2. ISelect 90K
2.6. Genetic Analyses
2.6.1. Construction of an Ultra-Dense Genetic Map
2.6.2. Linkage Analysis
2.6.3. QTL Gene Content
3. Results
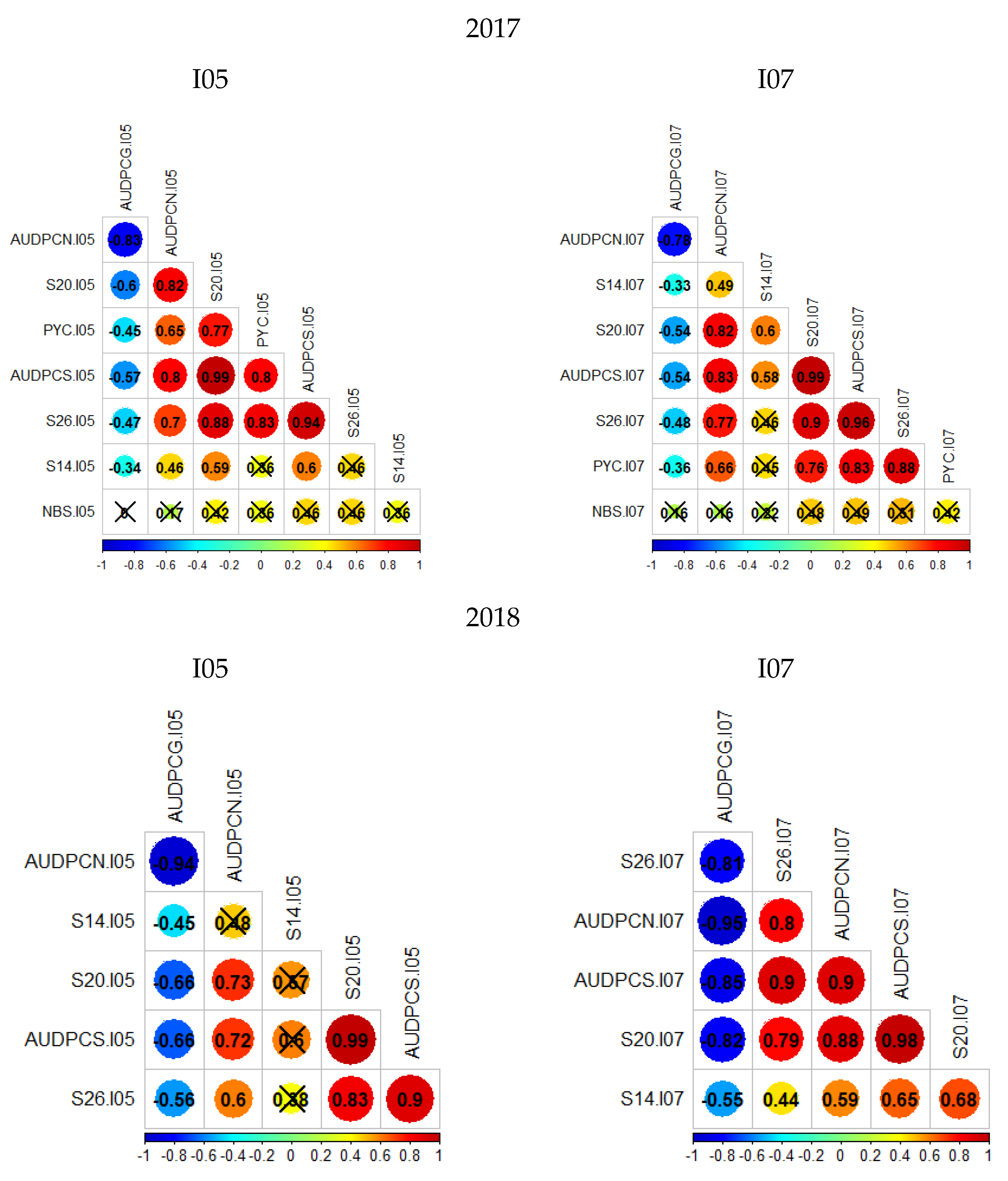
3.1. Description of Phenotypes
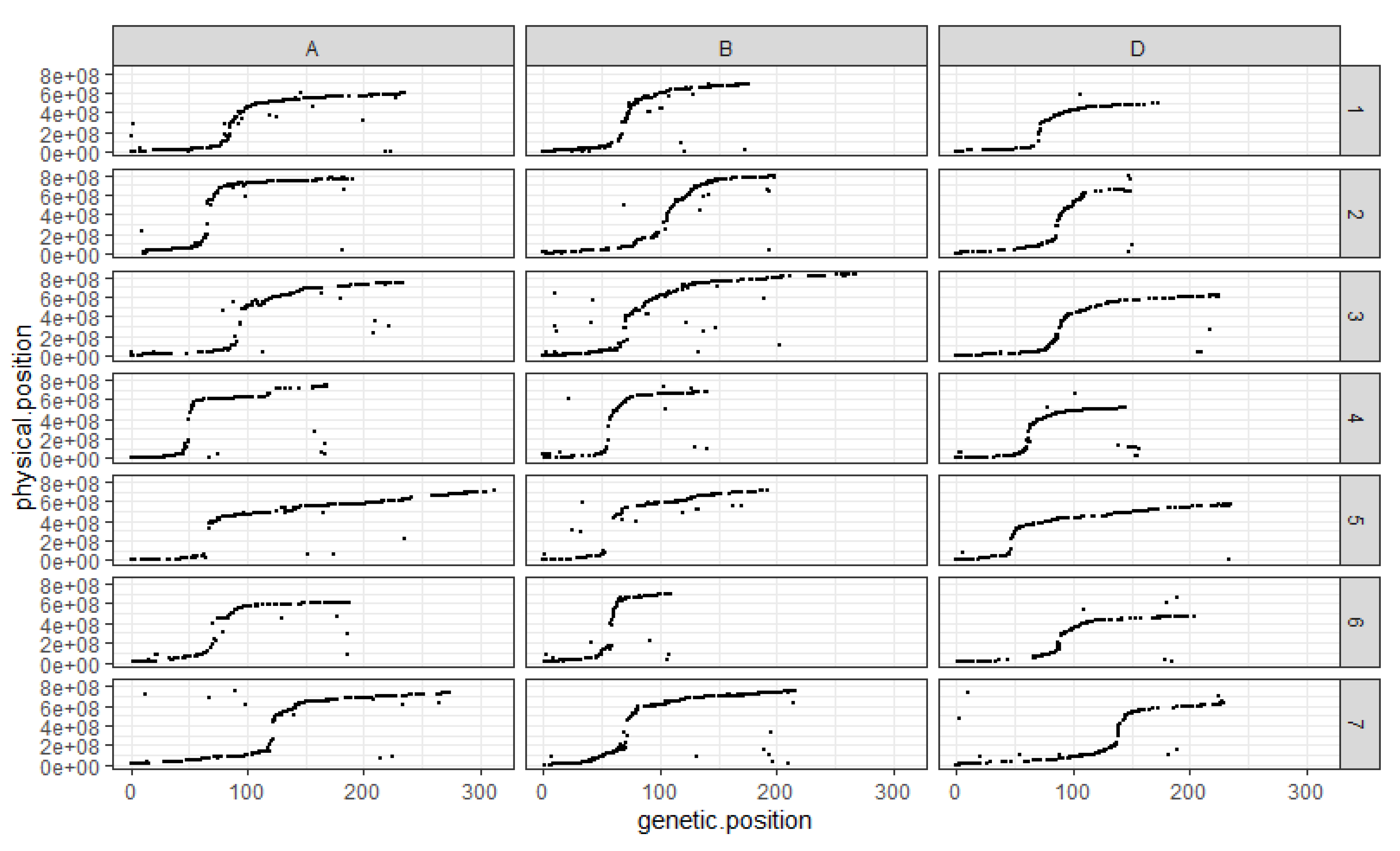
3.2. An Ultra-Dense Genetic Linkage Map
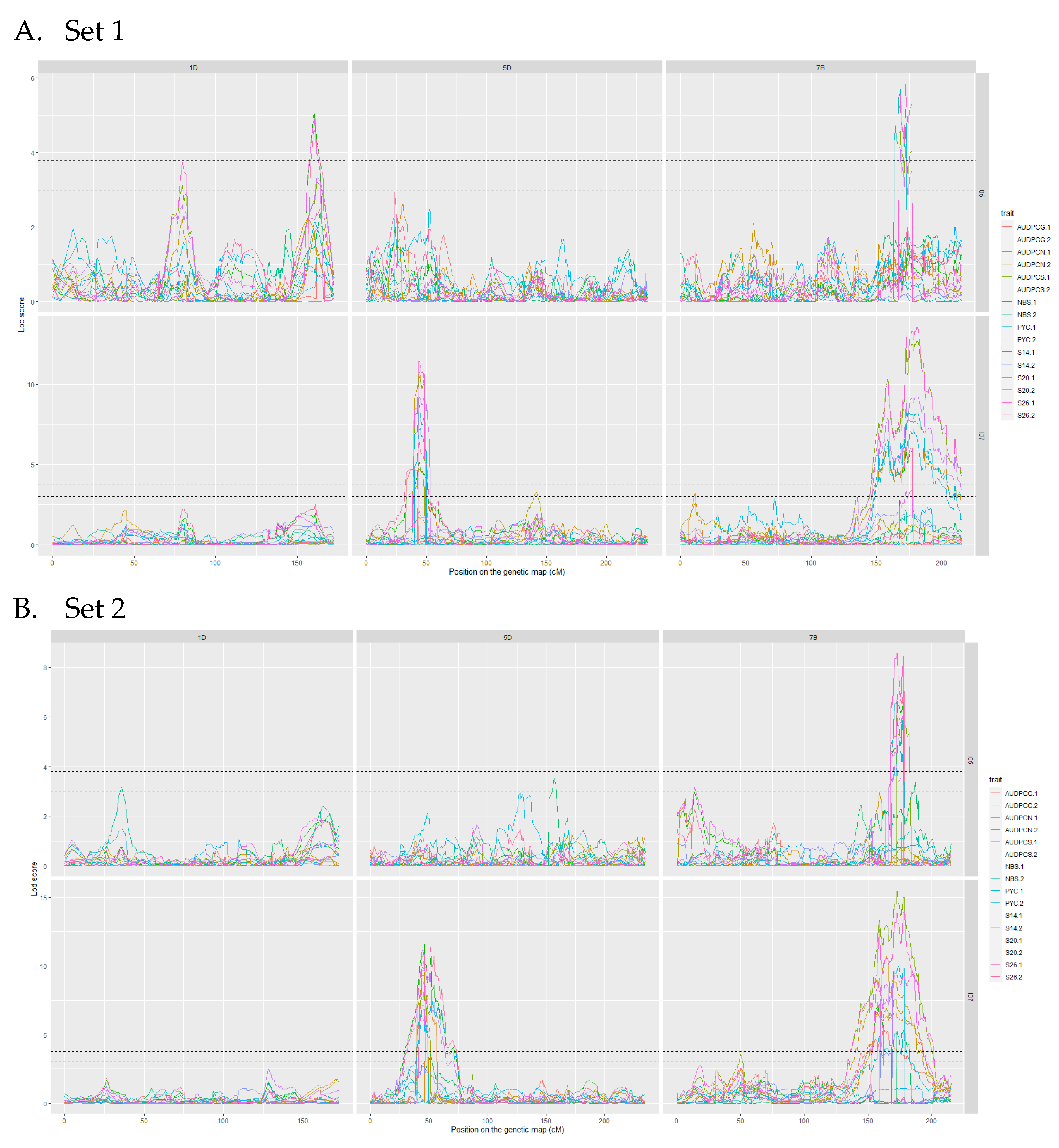
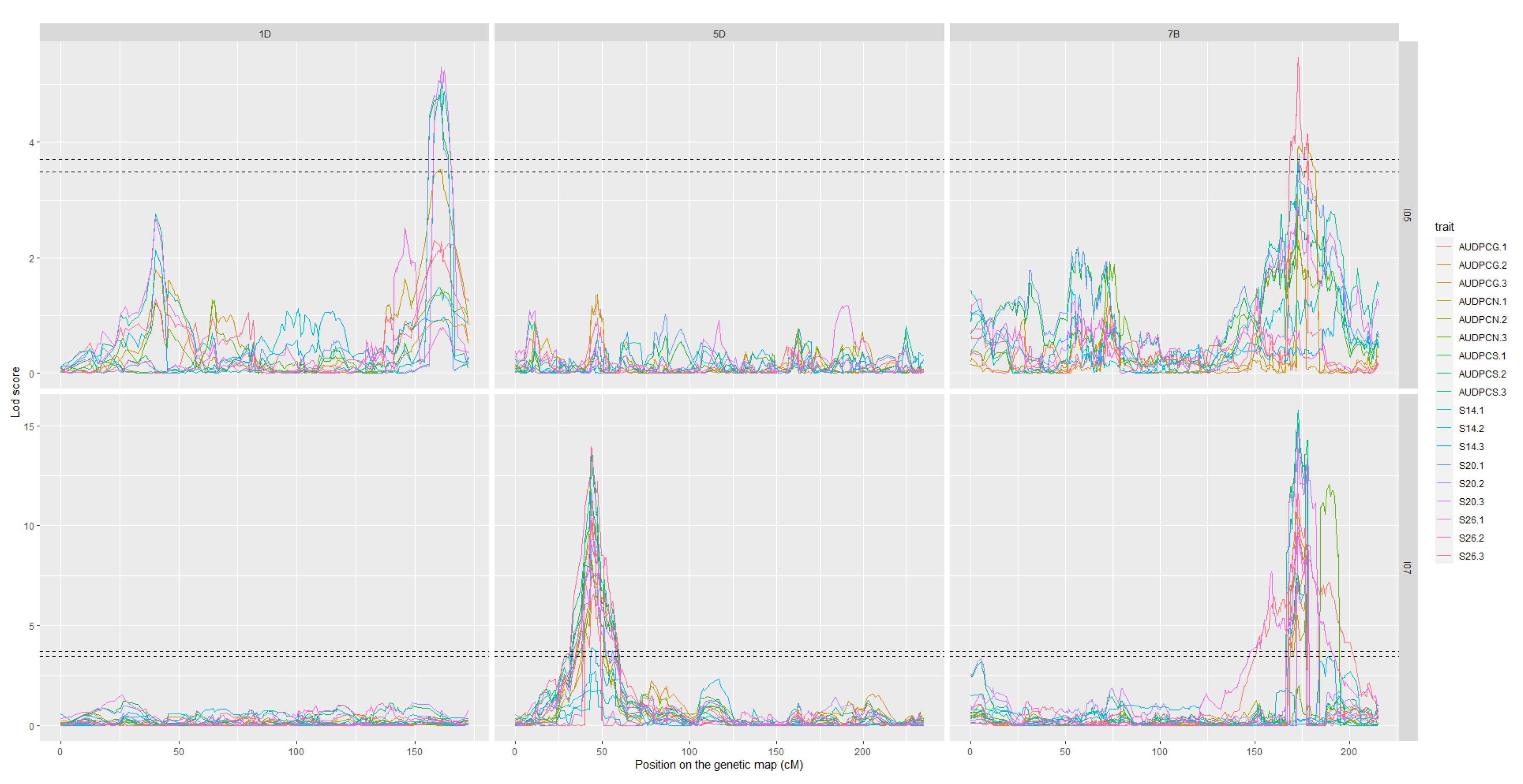
3.3. Mapping QTL for Resistance
3.4. Gene Content of the QTL
4. Discussion
4.1. An Ultra-Dense Genetic Map Built from Two SNP Arrays
4.2. Phenotypic Traits Involved in the Resistance of Renan to STB
4.3. Quantitative Resistance Durability Is a Multi-Layered Issue
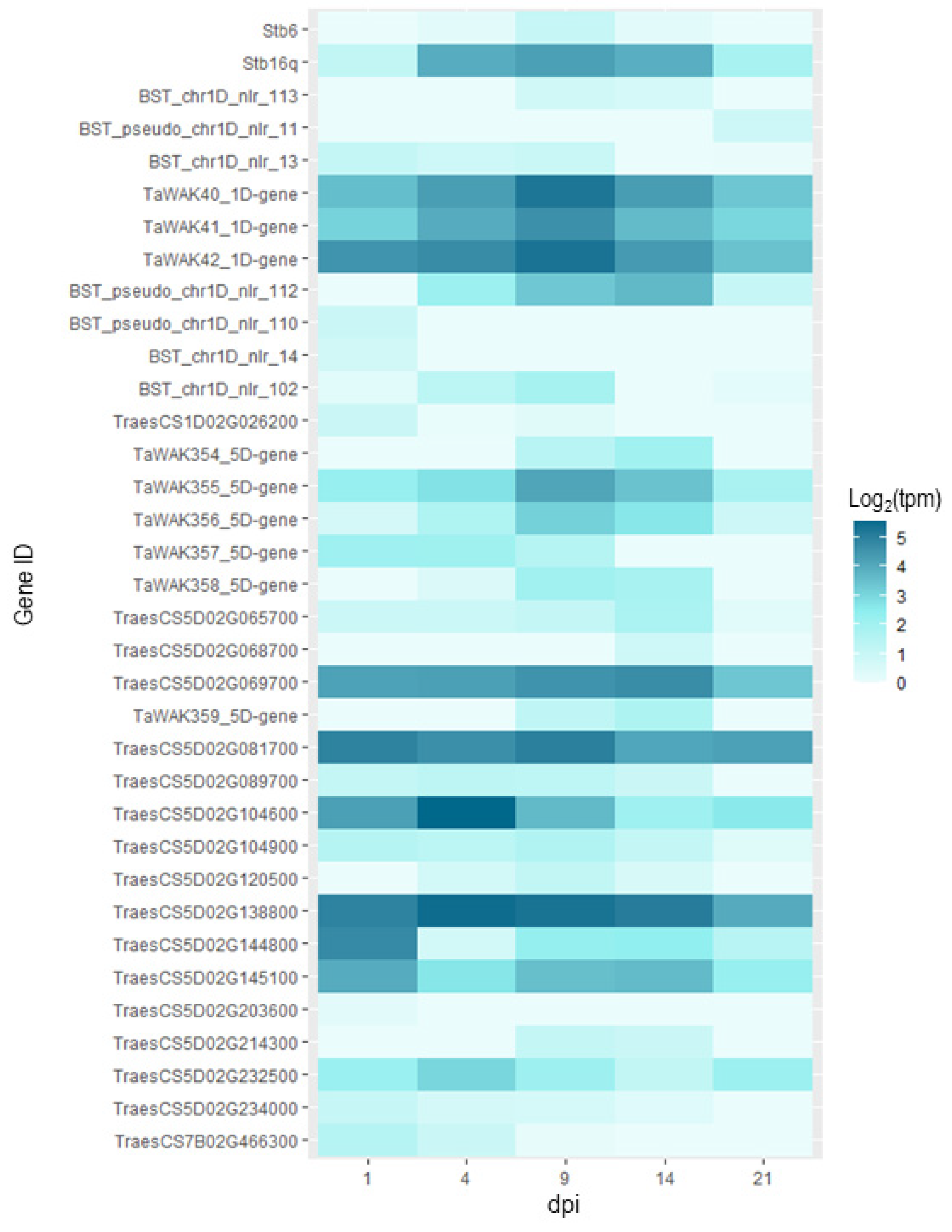
4.4. Molecular Mechanisms Underlying Resistance QTL
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Eyal, Z.; Scharen, A.L.; Prescott, J.M.; van Ginkel, M. The Septoria Diseases of Wheat: Concepts and Methods of Disease Management; CIMMYT: México-Veracruz, Mexico, 1987; 52p, ISBN 978-968-6127-06-5. [Google Scholar]
- Fones, H.; Gurr, S. The impact of Septoria tritici Blotch disease on wheat: An EU perspective. Fungal Genet. Biol. 2015, 79, 3–7. [Google Scholar] [CrossRef] [Green Version]
- O’Driscoll, A.; Kildea, S.; Doohan, F.; Spink, J.; Mullins, E. The wheat–Septoria conflict: A new front opening up? Trends Plant Sci. 2014, 19, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Niks, R.E.; Qi, X.; Marcel, T.C. Quantitative Resistance to Biotrophic Filamentous Plant Pathogens: Concepts, Misconceptions, and Mechanisms. Annu. Rev. Phytopathol. 2015, 53, 445–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dmochowska-Boguta, M.; Kloc, Y.; Zielezinski, A.; Werecki, P.; Nadolska-Orczyk, A.; Karlowski, W.M.; Orczyk, W. TaWAK6 encoding wall-associated kinase is involved in wheat resistance to leaf rust similar to adult plant resistance. PLoS ONE 2020, 15, e0227713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, E.; Kim, B.-S.; Iyer-Pascuzzi, A.S. Mechanisms of quantitative disease resistance in plants. Semin. Cell Dev. Biol. 2016, 56, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Uauy, C.; Distelfeld, A.; Blechl, A.; Epstein, L.; Chen, X.; Sela, H.; Fahima, T.; Dubcovsky, J. A Kinase-START Gene Confers Temperature-Dependent Resistance to Wheat Stripe Rust. Science 2009, 323, 1357–1360. [Google Scholar] [CrossRef] [Green Version]
- Fukuoka, S.; Okuno, K. QTL analysis and mapping of pi21, a recessive gene for field resistance to rice blast in Japanese upland rice. Theor. Appl. Genet. 2001, 103, 185–190. [Google Scholar] [CrossRef]
- Fukuoka, S.; Saka, N.; Koga, H.; Ono, K.; Shimizu, T.; Ebana, K.; Hayashi, N.; Takahashi, A.; Hirochika, H.; Okuno, K.; et al. Loss of Function of a Proline-Containing Protein Confers Durable Disease Resistance in Rice. Science 2009, 325, 998–1001. [Google Scholar] [CrossRef]
- Gadaleta, A.; Colasuonno, P.; Giove, S.L.; Blanco, A.; Giancaspro, A. Map-based cloning of QFhb.mgb-2A identifies a WAK2 gene responsible for Fusarium Head Blight resistance in wheat. Sci. Rep. 2019, 9, 6929. [Google Scholar] [CrossRef] [Green Version]
- Jiang, G.; Liu, D.; Yin, D.; Zhou, Z.; Shi, Y.; Li, C.; Zhu, L.; Zhai, W. A Rice NBS-ARC Gene Conferring Quantitative Resistance to Bacterial Blight Is Regulated by a Pathogen Effector-Inducible miRNA. Mol. Plant 2020, 13, 1752–1767. [Google Scholar] [CrossRef]
- Krattinger, S.G.; Lagudah, E.S.; Spielmeyer, W.; Singh, R.P.; Huerta-Espino, J.; McFadden, H.; Bossolini, E.; Selter, L.L.; Keller, B. A Putative ABC Transporter Confers Durable Resistance to Multiple Fungal Pathogens in Wheat. Science 2009, 323, 1360–1363. [Google Scholar] [CrossRef] [Green Version]
- Manosalva, P.M.; Davidson, R.M.; Liu, B.; Zhu, X.; Hulbert, S.H.; Leung, H.; Leach, J.E. A Germin-Like Protein Gene Family Functions as a Complex Quantitative Trait Locus Conferring Broad-Spectrum Disease Resistance in Rice. Plant Physiol. 2009, 149, 286–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Subedi, S.; De Vries, H.; Doornenbal, P.; Vels, A.; Hensel, G.; Kumlehn, J.; Johnston, P.A.; Qi, X.; Blilou, I.; et al. Orthologous receptor kinases quantitatively affect the host status of barley to leaf rust fungi. Nat. Plants 2019, 5, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.K.; Chartrain, L.; Lasserre-Zuber, P.; Saintenac, C. Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genet. Biol. 2015, 79, 33–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; McDonald, M.C.; Solomon, P.S.; Milgate, A.W. Genetic mapping of Stb19, a new resistance gene to Zymoseptoria tritici in wheat. Theor. Appl. Genet. 2018, 131, 2765–2773. [Google Scholar] [CrossRef]
- Vagndorf, N.; Nielsen, N.H.; Edriss, V.; Andersen, J.R.; Orabi, J.; Jørgensen, L.N.; Jahoor, A. Genomewide association study reveals novel quantitative trait loci associated with resistance towards Septoria tritici blotch in North European winter wheat. Plant Breed. 2017, 136, 474–482. [Google Scholar] [CrossRef]
- Gerard, G.S.; Börner, A.; Lohwasser, U.; Simon, M.R. Genome-wide association mapping of genetic factors controlling Septoria tritici blotch resistance and their associations with plant height and heading date in wheat. Euphytica 2017, 213, 27. [Google Scholar] [CrossRef]
- Karlstedt, F.; Kopahnke, D.; Perovic, D.; Jacobi, A.; Pillen, K.; Ordon, F. Mapping of quantitative trait loci (QTL) for resistance against Zymoseptoria tritici in the winter spelt wheat accession HTRI1410 (Triticum aestivum subsp. spelta). Euphytica 2019, 215, 108. [Google Scholar] [CrossRef]
- Odilbekov, F.; He, X.; Armoniené, R.; Saripella, G.V.; Henriksson, T.; Singh, P.K.; Chawade, A. QTL Mapping and Transcriptome Analysis to Identify Differentially Expressed Genes Induced by Septoria Tritici Blotch Disease of Wheat. Agronomy 2019, 9, 510. [Google Scholar] [CrossRef] [Green Version]
- Yates, S.; Mikaberidze, A.; Krattinger, S.G.; Abrouk, M.; Hund, A.; Yu, K.; Studer, B.; Fouche, S.; Meile, L.; Pereira, D.; et al. Precision Phenotyping Reveals Novel Loci for Quantitative Resistance to Septoria Tritici Blotch. Plant Phenomics 2019, 2019, 3285904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riaz, A.; KockAppelgren, P.; Hehir, J.G.; Kang, J.; Meade, F.; Cockram, J.; Milbourne, D.; Spink, J.; Mullins, E.; Byrne, S. Genetic Analysis Using a Multi-Parent Wheat Population Identifies Novel Sources of Septoria Tritici Blotch Resistance. Genes 2020, 11, 887. [Google Scholar] [CrossRef] [PubMed]
- Saintenac, C.; Lee, W.-S.; Cambon, F.; Rudd, J.J.; King, R.C.; Marande, W.; Powers, S.J.; Bergès, H.; Phillips, A.L.; Uauy, C.; et al. Wheat receptor-kinase-like protein Stb6 controls gene-for-gene resistance to fungal pathogen Zymoseptoria tritici. Nat. Genet. 2018, 50, 368–374. [Google Scholar] [CrossRef] [PubMed]
- Saintenac, C.; Cambon, F.; Aouini, L.; Verstappen, E.; Ghaffary, S.M.T.; Poucet, T.; Marande, W.; Berges, H.; Xu, S.; Jaouannet, M.; et al. A wheat cysteine-rich receptor-like kinase confers broad-spectrum resistance against Septoria tritici blotch. Nat. Commun. 2021, 12, 433. [Google Scholar] [CrossRef]
- Zhong, Z.; Marcel, T.C.; Hartmann, F.E.; Ma, X.; Plissonneau, C.; Zala, M.; Ducasse, A.; Confais, J.; Compain, J.; Lapalu, N.; et al. A small secreted protein in Zymoseptoria tritici is responsible for avirulence on wheat cultivars carrying the Stb6 resistance gene. New Phytol. 2017, 214, 619–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doré, C.; Varoquaux, F. Histoire et Amélioration de Cinquante Plantes Cultivées; Editions Quae: Versailles, France, 2006; 812p, ISBN 2-7380-1215-9. [Google Scholar]
- Doussinault, G.; Pavoine, M.-T.; Jaudeau, B.; Jahier, J. Évolution de la Variabilité Génétique chez le Blé. Doss. Environ. INRA 2001, 21, 91–104. [Google Scholar]
- Hanzalova, A.; Dumalasova, V.; Sumikova, T.; Bartos, P. Rust Resistance of the French Wheat Cultivar Renan. Czech J. Genet. Plant Breed 2007, 43, 53–60. [Google Scholar] [CrossRef] [Green Version]
- Dedryver, F.; Paillard, S.; Mallard, S.; Robert, O.; Trottet, M.; Nègre, S.; Verplancke, G.; Jahier, J. Characterization of Genetic Components Involved in Durable Resistance to Stripe Rust in the Bread Wheat ‘Renan’. Phytopathology 2009, 99, 968–973. [Google Scholar] [CrossRef]
- Rolland, B.; Fontaine, L.; Mailliard, A.; Gardet, O.; Heumez, E.; Walczak, P.; Le Campion, A.; Oury, F.-X. From selection to cultivation with the support of all stakeholders: The first registration in France of two winter bread wheat varieties after value for cultivation and use evaluation in organic farming systems. Org. Agric. 2017, 7, 73–81. [Google Scholar] [CrossRef]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the bread wheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The International Wheat Genome Sequencing Consortium (IWGSC); Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, eaar7191. [Google Scholar] [CrossRef] [Green Version]
- Zhu, T.; Wang, L.; Rimbert, H.; Rodriguez, J.C.; Deal, K.R.; De Oliveira, R.; Choulet, F.; Keeble-Gagnère, G.; Tibbits, J.; Rogers, J.; et al. Optical maps refine the bread wheat Triticum aestivum cv. Chinese Spring genome assembly. Plant J. 2021, 107, 303–314. [Google Scholar] [CrossRef]
- Uauy, C. Wheat genomics comes of age. Curr. Opin. Plant Biol. 2017, 36, 142–148. [Google Scholar] [CrossRef]
- Sears, E.R.; Miller, T.E. The History of Chinese Spring Wheat. Cereal Res. Commun. 1985, 13, 261–263. [Google Scholar]
- Qi, X.; Jiang, G.; Chen, W.; Niks, R.E.; Stam, P.; Lindhout, P. Isolate-specific QTLs for partial resistance to Puccinia hordei in barley. Theor. Appl. Genet. 1999, 99, 877–884. [Google Scholar] [CrossRef]
- González, A.M.; Marcel, T.C.; Niks, R.E. Evidence for a Minor Gene–for–Minor Gene Interaction Explaining Nonhypersensitive Polygenic Partial Disease Resistance. Phytopathology 2012, 102, 1086–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arru, L.; Francia, E.; Pecchioni, N. Isolate-specific QTLs of resistance to leaf stripe (Pyrenophora graminea) in the ‘Steptoe’ × ‘Morex’ spring barley cross. Theor. Appl. Genet. 2003, 106, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Leonards-Schippers, C.; Gieffers, W.; Schäfer-Pregl, R.; Ritter, E.; Knapp, S.J.; Salamini, F.; Gebhardt, C. Quantitative resistance to Phytophthora infestans in potato: A case study for QTL mapping in an allogamous plant species. Genetics 1994, 137, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Caranta, C.; Lefebvre, V.; Palloix, A. Polygenic Resistance of Pepper to Potyviruses Consists of a Combination of Isolate-Specific and Broad-Spectrum Quantitative Trait Loci. Mol. Plant-Microbe Interact. 1997, 10, 872–878. [Google Scholar] [CrossRef] [Green Version]
- Shaw, M.W. Interacting effects of interrupted humid periods and light on infection of wheat leaves by Mycosphaerella graminicola (Septoria tritici). Plant Pathol. 1991, 40, 595–607. [Google Scholar] [CrossRef]
- Boixel, A.-L.; Gélisse, S.; Marcel, T.C.; Suffert, F. Differential Tolerance to Changes in Moisture Regime during Early Infection Stages in the Fungal Pathogen Zymoseptoria Tritici. bioRxiv 2019, 867572. [Google Scholar] [CrossRef] [Green Version]
- Kema, G.H.J.; Yu, D.; Rijkenberg, F.H.J.; Shaw, M.W.; Baayen, R.P. Histology of the Pathogenesis of Mycosphaerella Graminicola in Wheat. Phytopathology 1996, 86, 777–786. [Google Scholar] [CrossRef]
- Ouaja, M.; Aouini, L.; Bahri, B.; Ferjaoui, S.; Medini, M.; Marcel, T.C.; Hamza, S. Identification of valuable sources of resistance to Zymoseptoria tritici in the Tunisian durum wheat landraces. Eur. J. Plant Pathol. 2020, 156, 647–661. [Google Scholar] [CrossRef]
- Stewart, E.L.; McDonald, B.A. Measuring Quantitative Virulence in the Wheat Pathogen Zymoseptoria tritici Using High-Throughput Automated Image Analysis. Phytopathology 2014, 104, 985–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, E.L.; Hagerty, C.H.; Mikaberidze, A.; Mundt, C.C.; Zhong, Z.; McDonald, B.A. An Improved Method for Measuring Quantitative Resistance to the Wheat Pathogen Zymoseptoria tritici Using High-Throughput Automated Image Analysis. Phytopathology 2016, 106, 782–788. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Palmer, C.-L.; Skinner, W. Mycosphaerella graminicola: Latent infection, crop devastation and genomics. Mol. Plant Pathol. 2002, 3, 63–70. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienne, Austria, 2019. [Google Scholar]
- Danguy des Déserts, A.; Bouchet, S.; Sourdille, P.; Servin, B. Evolution of recombination landscapes in diverging populations of bread wheat. Genome Biol. Evol. 2021, 13, evab152. [Google Scholar] [CrossRef]
- Kitt, J.; Danguy des Déserts, A.; Bouchet, S.; Servin, B.; Rimbert, H.; De Oliveira, R.; Choulet, F.; Balfourier, F.; Sourdille, P.; Paux, E. Genotyping of 4506 Bread Wheat Accessions with the TaBW410K SNP Array. Zenodo 2021. [Google Scholar] [CrossRef]
- Wang, S.; Wong, D.; Forrest, K.; Allen, A.; Chao, S.; Huang, B.E.; Maccaferri, M.; Salvi, S.; Milner, S.G.; Cattivelli, L.; et al. Characterization of polyploid wheat genomic diversity using a high-density 90 000 single nucleotide polymorphism array. Plant Biotechnol. J. 2014, 12, 787–796. [Google Scholar] [CrossRef] [Green Version]
- Ronin, Y.I.; Mester, D.I.; Minkov, D.G.; Akhunov, E.; Korol, A.B. Building Ultra-High-Density Linkage Maps Based on Efficient Filtering of Trustable Markers. Genetics 2017, 206, 1285–1295. [Google Scholar] [CrossRef] [Green Version]
- Kosambi, D.D. The Estimation of Map Distances from Recombination Values. In D.D. Kosambi: Selected Works in Mathematics and Statistics; Ramaswamy, R., Ed.; Springer India: New Delhi, India, 2016; pp. 125–130. ISBN 978-81-322-3676-4. [Google Scholar]
- Voorrips, R.E. MapChart: Software for the Graphical Presentation of Linkage Maps and QTLs. J. Hered. 2002, 93, 77–78. [Google Scholar] [CrossRef] [Green Version]
- Broman, K.W.; Wu, H.; Sen, Ś.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrill, P.; Ramirez-Gonzalez, R.; Uauy, C. expVIP: A Customizable RNA-seq Data Analysis and Visualization Platform. Plant Physiol. 2016, 170, 2172–2186. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-González, R.H.; Borrill, P.; Lang, D.; Harrington, S.A.; Brinton, J.; Venturini, L.; Davey, M.; Jacobs, J.; van Ex, F.; Pasha, A.; et al. The transcriptional landscape of polyploid wheat. Science 2018, 361, eaar6089. [Google Scholar] [CrossRef] [Green Version]
- Rudd, J.J.; Kanyuka, K.; Hassani-Pak, K.; Derbyshire, M.; Andongabo, A.; Devonshire, J.; Lysenko, A.; Saqi, M.; Desai, N.M.; Powers, S.J.; et al. Transcriptome and Metabolite Profiling of the Infection Cycle of Zymoseptoria tritici on Wheat Reveals a Biphasic Interaction with Plant Immunity Involving Differential Pathogen Chromosomal Contributions and a Variation on the Hemibiotrophic Lifestyle Definition. Plant Physiol. 2015, 167, 1158–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartrain, L.; Brading, P.A.; Brown, J.K.M. Presence of the Stb6 gene for resistance to septoria tritici blotch (Mycosphaerella graminicola) in cultivars used in wheat-breeding programmes worldwide. Plant Pathol. 2005, 54, 134–143. [Google Scholar] [CrossRef]
- Kourelis, J.; Van Der Hoorn, R.A.L. Defended to the Nines: 25 Years of Resistance Gene Cloning Identifies Nine Mechanisms for R Protein Function. Plant Cell 2018, 30, 285–299. [Google Scholar] [CrossRef] [Green Version]
- Steinberg, G. Cell biology of Zymoseptoria tritici: Pathogen cell organization and wheat infection. Fungal Genet. Biol. 2015, 79, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Badaeva, E.D.; Dedkova, O.S.; Gay, G.; Pukhalskyi, V.A.; Zelenin, A.V.; Bernard, S. Chromosomal rearrangements in wheat: Their types and distribution. Genome 2007, 50, 907–926. [Google Scholar] [CrossRef] [PubMed]
- Broderick, J.B. Coenzymes and Cofactors. In eLS; American Cancer Society: Atlanta, GA, USA, 2001; ISBN 978-0-470-01590-2. [Google Scholar]
- Rimbert, H.; Darrier, B.; Navarro, J.; Kitt, J.; Choulet, F.; Leveugle, M.; Duarte, J.; Rivière, N.; Eversole, K.; Le Gouis, J.; et al. High throughput SNP discovery and genotyping in hexaploid wheat. PLoS ONE 2018, 13, e0186329. [Google Scholar] [CrossRef] [Green Version]
- Wen, W.; He, Z.; Gao, F.; Liu, J.; Jin, H.; Zhai, S.; Qu, Y.; Xia, X. A High-Density Consensus Map of Common Wheat Integrating Four Mapping Populations Scanned by the 90K SNP Array. Front. Plant Sci. 2017, 8, 1389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, W.; Baenziger, P.S.; Belamkar, V.; Guttieri, M.J.; Venegas, J.P.; Easterly, A.; Sallam, A.; Poland, J. Genotyping-by-Sequencing Derived High-Density Linkage Map and its Application to QTL Mapping of Flag Leaf Traits in Bread Wheat. Sci. Rep. 2017, 7, 16394. [Google Scholar] [CrossRef]
- Ladejobi, O.; Mackay, I.J.; Poland, J.; Praud, S.; Hibberd, J.M.; Bentley, A.R. Reference Genome Anchoring of High-Density Markers for Association Mapping and Genomic Prediction in European Winter Wheat. Front. Plant Sci. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, W.C.; Haueisen, J.; Stukenbrock, E.H. Dissecting the Biology of the Fungal Wheat Pathogen Zymoseptoria tritici: A Laboratory Workflow. Curr. Protoc. Microbiol. 2020, 59, e128. [Google Scholar] [CrossRef]
- Riaz, A.; Hickey, L.T. Rapid Phenotyping Adult Plant Resistance to Stem Rust in Wheat Grown under Controlled Conditions. In Wheat Rust Diseases: Methods and Protocols; Periyannan, S., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2017; pp. 183–196. ISBN 978-1-4939-7249-4. [Google Scholar]
- Habig, M.; Quade, J.; Stukenbrock, E.H. Forward Genetics Approach Reveals Host Genotype-Dependent Importance of Accessory Chromosomes in the Fungal Wheat Pathogen Zymoseptoria tritici. MBio 2017, 8, e01919-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boixel, A.-L.; Delestre, G.; Legeay, J.; Chelle, M.; Suffert, F. Phenotyping Thermal Responses of Yeasts and Yeast-like Microorganisms at the Individual and Population Levels: Proof-of-Concept, Development and Application of an Experimental Framework to a Plant Pathogen. Microb. Ecol. 2019, 78, 42–56. [Google Scholar] [CrossRef]
- Maupetit, A.; Larbat, R.; Pernaci, M.; Andrieux, A.; Guinet, C.; Boutigny, A.-L.; Fabre, B.; Frey, P.; Halkett, F. Defense Compounds Rather Than Nutrient Availability Shape Aggressiveness Trait Variation Along a Leaf Maturity Gradient in a Biotrophic Plant Pathogen. Front. Plant Sci. 2018, 9, 1396. [Google Scholar] [CrossRef]
- Thomas, C.; Mabon, R.; Andrivon, D.; Val, F. The Effectiveness of Induced Defense Responses in a Susceptible Potato Genotype Depends on the Growth Rate of Phytophthora infestans. Mol. Plant-Microbe Interact. 2019, 32, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Dutt, A.; Andrivon, D.; Jumel, S.; Le Roy, G.; Baranger, A.; Leclerc, M.; Le May, C. Life history traits and trade-offs between two species of the ascochyta blight disease complex of pea. Plant Pathol. 2020, 69, 1108–1124. [Google Scholar] [CrossRef]
- Pilet-Nayel, M.-L.; Moury, B.; Caffier, V.; Montarry, J.; Kerlan, M.-C.; Fournet, S.; Durel, C.-E.; Delourme, R. Quantitative Resistance to Plant Pathogens in Pyramiding Strategies for Durable Crop Protection. Front. Plant Sci. 2017, 8, 1838. [Google Scholar] [CrossRef] [Green Version]
- Parlevliet, J.E. Durability of resistance against fungal, bacterial and viral pathogens; present situation. Euphytica 2002, 124, 147–156. [Google Scholar] [CrossRef]
- Harrison, B.D. Virus variation in relation to resistance-breaking in plants. Euphytica 2002, 124, 181–192. [Google Scholar] [CrossRef]
- Lindhout, P. The perspectives of polygenic resistance in breeding for durable disease resistance. Euphytica 2002, 124, 217–226. [Google Scholar] [CrossRef]
- Palloix, A.; Ayme, V.; Moury, B. Durability of plant major resistance genes to pathogens depends on the genetic background, experimental evidence and consequences for breeding strategies. New Phytol. 2009, 183, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Quenouille, J.; Paulhiac, E.; Moury, B.; Palloix, A. Quantitative trait loci from the host genetic background modulate the durability of a resistance gene: A rational basis for sustainable resistance breeding in plants. Heredity 2014, 112, 579–587. [Google Scholar] [CrossRef] [Green Version]
- Van, A.L.; Caffier, V.; Lasserre-Zuber, P.; Chauveau, A.; Brunel, D.; Le Cam, B.; Durel, C. Differential selection pressures exerted by host resistance quantitative trait loci on a pathogen population: A case study in an apple × Venturia inaequalis pathosystem. New Phytol. 2013, 197, 899–908. [Google Scholar] [CrossRef] [PubMed]
- Montarry, J.; Cartier, E.; Jacquemond, M.; Palloix, A.; Moury, B. Virus adaptation to quantitative plant resistance: Erosion or breakdown? J. Evol. Biol. 2012, 25, 2242–2252. [Google Scholar] [CrossRef]
- Caffier, V.; Lasserre-Zuber, P.; Giraud, M.; Lascostes, M.; Stievenard, R.; Lemarquand, A.; van de Weg, E.; Expert, P.; Denancé, C.; Didelot, F.; et al. Erosion of quantitative host resistance in the apple × Venturia inaequalis pathosystem. Infect. Genet. Evol. 2014, 27, 481–489. [Google Scholar] [CrossRef]
- Lehman, J.S.; Shaner, G. Selection of Populations of Puccinia recondita f. sp. tritici for Shortened Latent Period on a Partially Resistant Wheat Cultivar. Phytopathology 1997, 87, 170–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delmotte, F.; Mestre, P.; Schneider, C.; Kassemeyer, H.-H.; Kozma, P.; Richart-Cervera, S.; Rouxel, M.; Delière, L. Rapid and multiregional adaptation to host partial resistance in a plant pathogenic oomycete: Evidence from European populations of Plasmopara viticola, the causal agent of grapevine downy mildew. Infect. Genet. Evol. 2014, 27, 500–508. [Google Scholar] [CrossRef]
- Poland, J.A.; Balint-Kurti, P.J.; Wisser, R.J.; Pratt, R.C.; Nelson, R.J. Shades of gray: The world of quantitative disease resistance. Trends Plant Sci. 2009, 14, 21–29. [Google Scholar] [CrossRef]
- Seah, S.; Bariana, H.; Jahier, J.; Sivasithamparam, K.; Lagudah, E.S. The introgressed segment carrying rust resistance genes Yr17, Lr37 and Sr38 in wheat can be assayed by a cloned disease resistance gene-like sequence. Theor. Appl. Genet. 2001, 102, 600–605. [Google Scholar] [CrossRef]
- Bartos, P.; Ovesná, J.; Hanzalová, A.; Chrpová, J.; Dumalasová, V.; Škorpík, M.; Šíp, V. Presence of a Translocation from Aegilops ventricosa in Wheat Cultivars Registered in the Czech Republic. Czech J. Genet. Plant Breed. 2011, 40, 31–35. [Google Scholar] [CrossRef] [Green Version]
- Ambrozková, M.; Dedryver, F.; Dumalasová, V.; Hanzalová, A.; Bartos, P. Determination of the cluster of wheat rust resistance genes Yr17, Lr37, and Sr38 by a molecular marker. Plant Prot. Sci. 2002, 38, 41–45. [Google Scholar] [CrossRef] [Green Version]
- Gallais, A. Blé Renan: Un OGM Ignoré Très Utilisé Par L’agriculture Biologique. Available online: https://www.biotechnologies-vegetales.com/wp-content/uploads/2020/04/FicheInfo06-Ble-Renan-un-OGM-ignore-tres-utilise-par-l-agriculture-bio.pdf (accessed on 3 December 2021).
- Pasquariello, M.; Ham, J.; Burt, C.; Jahier, J.; Paillard, S.; Uauy, C.; Nicholson, P. The eyespot resistance genes Pch1 and Pch2 of wheat are not homoeoloci. Theor. Appl. Genet. 2016, 130, 91–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Xie, J.; Hu, J.; Qiu, D.; Liu, Z.; Li, J.; Li, M.; Zhang, H.; Yang, L.; Liu, H.; et al. Development of Molecular Markers Linked to Powdery Mildew Resistance Gene Pm4b by Combining SNP Discovery from Transcriptome Sequencing Data with Bulked Segregant Analysis (BSR-Seq) in Wheat. Front. Plant Sci. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, T.B.; Anderson, J.M.; Goodwin, S.B. Identification and Molecular Mapping of a Gene in Wheat Conferring Resistance to Mycosphaerella graminicola. Phytopathology 2003, 93, 1158–1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chartrain, L.; Berry, S.T.; Brown, J.K.M. Resistance of Wheat Line Kavkaz-K4500 L.6.A.4 to Septoria Tritici Blotch Controlled by Isolate-Specific Resistance Genes. Phytopathology 2005, 95, 664–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faris, J.D.; Zhang, Z.; Lu, H.; Lu, S.; Reddy, L.; Cloutier, S.; Fellers, J.P.; Meinhardt, S.W.; Rasmussen, J.B.; Xu, S.S.; et al. A Unique Wheat Disease Resistance-like Gene Governs Effector-Triggered Susceptibility to Necrotrophic Pathogens. Proc. Natl. Acad. Sci. USA 2010, 107, 13544–13549. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Particle Counter | Diameter | 0–8 µm |
| Size | 7–85 µm | |
| Circonference | 0–0.70 µm | |
| Grey value | 195–205 | |
| Luminous intensity | 7.5 | |
| Spacer thickness | 150 µm | |
| Resolution | Magnification | ×4 |
| Calibration | 0.47 µm.pixel−1 (1 pixel = 1.67 µm) | |
| Isolate Set | Trait | Statistical Significance of the Genotype 1 | Statistical Significance of the Replication 1 | MSg 2 | MSε 3 | Broad-Sense Heritability | Shapiro–Wilk Normality Test on Residuals | Independence of Residuals | Homoscedasticity Bartlett Test of Homogeneity of Variances |
|---|---|---|---|---|---|---|---|---|---|
| I05_2017/1 | S14 | *** | 27 | 27 | 0.00 | 0.00 | no | 0.04 | |
| S20 | *** | *** | 1094 | 444 | 0.42 | 0.11 | yes | 0.14 | |
| S26 | *** | *** | 877 | 364 | 0.41 | 0.12 | yes | 0.03 | |
| AUDPCG | * | * | 51,787 | 35,921 | 0.18 | 0.92 | yes | 0.05 | |
| AUDPCN | * | * | 77,482 | 55,907 | 0.16 | 0.85 | yes | 0.31 | |
| AUDPCS | *** | *** | 83,562 | 32,115 | 0.44 | 0.11 | yes | 0.26 | |
| PYC | *** | 21,769 | 9037 | 0.41 | 0.29 | yes | 0.29 | ||
| NBS | *** | 1,003,915 | 868,109 | 0.07 | 0.00 | yes | 0.05 | ||
| I07_2017/1 | S14 | ** | 2 | 2 | 0.04 | 0.00 | no | 0.00 | |
| S20 | *** | *** | 401 | 157 | 0.44 | 0.00 | yes | 0.85 | |
| S26 | *** | *** | 765 | 233 | 0.53 | 0.40 | yes | 0.98 | |
| AUDPCG | *** | 57,879 | 30,256 | 0.31 | 0.00 | no | 0.00 | ||
| AUDPCN | *** | *** | 92,012 | 25,943 | 0.56 | 0.95 | yes | 0.77 | |
| AUDPCS | *** | *** | 40,045 | 12,909 | 0.51 | 0.00 | yes | 0.97 | |
| PYC | *** | 29,153 | 8834 | 0.53 | 0.01 | no | 0.04 | ||
| NBS | *** | 1,865,055 | 630,731 | 0.49 | 0.00 | no | 0.00 | ||
| I05_2017/2 | S14 | *** | * | 14 | 7 | 0.34 | 0.00 | no | 0.00 |
| S20 | ** | . | 533 | 344 | 0.22 | 0.94 | yes | 0.54 | |
| S26 | ** | 665 | 378 | 0.28 | 0.40 | yes | 0.89 | ||
| AUDPCG | * | *** | 38,627 | 27,765 | 0.16 | 0.02 | no | 0.04 | |
| AUDPCN | * | *** | 62,573 | 42,711 | 0.19 | 0.93 | yes | 0.35 | |
| AUDPCS | ** | . | 44,817 | 26,387 | 0.26 | 0.74 | yes | 0.79 | |
| PYC | *** | *** | 22,651 | 12,737 | 0.28 | 0.97 | yes | 0.34 | |
| NBS | ** | *** | 816,528 | 495,995 | 0.24 | 0.00 | yes | 0.04 | |
| I07_2017/2 | S14 | *** | 31 | 13 | 0.42 | 0.00 | no | 0.00 | |
| S20 | *** | 1195 | 189 | 0.73 | 0.00 | no | 0.01 | ||
| S26 | *** | 1411 | 222 | 0.73 | 0.60 | yes | 0.33 | ||
| AUDPCG | *** | 79,008 | 28,924 | 0.46 | 0.16 | yes | 0.22 | ||
| AUDPCN | *** | 115,168 | 23,787 | 0.66 | 0.24 | yes | 0.01 | ||
| AUDPCS | *** | 102,594 | 13,479 | 0.77 | 0.00 | no | 0.03 | ||
| PYC | *** | *** | 59,415 | 18,267 | 0.53 | 0.82 | yes | 0.65 | |
| NBS | * | * | 1,341,693 | 906,931 | 0.19 | 0.00 | yes | 0.27 | |
| I05_2018 only replications 1 and 3 | S14 | ** | *** | 140 | 86 | 0.24 | 0.00 | no | 0.00 |
| S20 | *** | *** | 1649 | 747 | 0.38 | 0.28 | yes | 0.94 | |
| S26 | * | *** | 877 | 661 | 0.14 | 0.00 | no | 0.82 | |
| AUDPCG | *** | *** | 32,787 | 17,602 | 0.30 | 0.00 | no | 0.00 | |
| AUDPCN | *** | *** | 58,506 | 28,592 | 0.34 | 0.86 | yes | 0.26 | |
| AUDPCS | *** | *** | 115,164 | 50,508 | 0.39 | 0.66 | yes | 0.65 | |
| I07_2018 | S14 | *** | 43 | 22 | 0.25 | 0.00 | no | 0.00 | |
| S20 | *** | ** | 3061 | 368 | 0.71 | 0.00 | no | 0.00 | |
| S26 | *** | *** | 3270 | 477 | 0.66 | 0.00 | no | 0.00 | |
| AUDPCG | *** | 144,099 | 32,073 | 0.54 | 0.32 | yes | 0.02 | ||
| AUDPCN | *** | ** | 170,267 | 29,795 | 0.61 | 0.00 | yes | 0.00 | |
| AUDPCS | *** | *** | 242,232 | 25,587 | 0.74 | 0.01 | yes | 0.00 |
| Chromosome | Number of SNP | Number of Genetic Bins | Map Length (cM) | Marker Density (Markers/cM) | |
|---|---|---|---|---|---|
| A—genome | 1 | 10,479 | 358 | 234.44 | 44.7 |
| 2 | 11,716 | 288 | 189.72 | 61.8 | |
| 3 | 8413 | 302 | 233.28 | 36.1 | |
| 4 | 8465 | 203 | 170.24 | 49.7 | |
| 5 | 6723 | 411 | 312.17 | 21.5 | |
| 6 | 7816 | 226 | 189.03 | 41.3 | |
| 7 | 10,051 | 378 | 273.38 | 36.7 | |
| total A | 63,663 | 2167 | 1602.26 | 39.7 | |
| B—genome | 1 | 10,503 | 309 | 176.4 | 59.5 |
| 2 | 9158 | 298 | 197.98 | 46.3 | |
| 3 | 12,016 | 374 | 268.03 | 44.8 | |
| 4 | 5888 | 180 | 140.45 | 41.9 | |
| 5 | 5019 | 234 | 192.98 | 26 | |
| 6 | 10,210 | 173 | 109.66 | 93.1 | |
| 7 | 7803 | 336 | 215.35 | 36.2 | |
| total B | 60,597 | 1905 | 1300.85 | 46.6 | |
| D—genome | 1 | 3609 | 157 | 172.81 | 20.9 |
| 2 | 4601 | 134 | 150.27 | 30.6 | |
| 3 | 3512 | 225 | 225.25 | 15.6 | |
| 4 | 2131 | 174 | 156.99 | 13.6 | |
| 5 | 2581 | 203 | 234.75 | 11 | |
| 6 | 4180 | 183 | 204.6 | 20.4 | |
| 7 | 3946 | 211 | 228.97 | 17.2 | |
| total D | 24,560 | 1287 | 1373.64 | 17.9 | |
| Total | 148,820 | 5357 | 4276.75 | 34.8 | |
| QTL.2017 | Number of Detections | r2 Max (%) | Mean r2 (%) | Peak Marker Associated with r2 Max | Parent Carrying the Resistance Allele | Traits | Detected with |
|---|---|---|---|---|---|---|---|
| Qstb.renan-1D | 3 | 7.5 | 6 | cfn1317667_410K_1DS | Renan | S20, S26, AUDPCS | I05 |
| Qstb.renan-5D | 22 | 35.5 | 26 | cfn2823104_410K_5DS | Renan | S20, S26, AUDPCG, AUDPCN, AUDPCS, PYC | I07 |
| Qstb.renan-7B | 37 | 32 | 20 | cfn0449267_410K_7BL | Renan | S14, S20, S26, AUDPCG, AUDPCN, AUDPCS, PYC, NBS | I05 and I07 |
| QTL.2018 | Number of Detections | r2 Max (%) | Mean r2 (%) | Peak Marker Associated with r2 Max | Parent Carrying the Resistance Allele | Traits | Detected with |
|---|---|---|---|---|---|---|---|
| Qstb.renan-1D | 5 | 15 | 13.5 | cfn1315024_410K_1DS | Renan | S20, AUDPCN, AUDPCS | I05 |
| Qstb.renan-5D | 18 | 21.5 | 15.5 | cfn2827993_410K_5DS | Renan | S14, S20, S26, AUDPCG, AUDPCN, AUDPCS | I07 |
| Qstb.renan-7B | 22 | 38 | 21 | cfn0916416_410K_7BL | Renan | S14, S20, S26, AUDPCG, AUDPCN, AUDPCS | I05 and I07 |
| QTL | Gene.ID | RefSeq v1.1 ID | Start (bp) | Stop (bp) | Annotation |
|---|---|---|---|---|---|
| Qstb-renan-1D | BST_chr1D_nlr_115 | TraesCS1D02G015500 | 7277369 | 7280463 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_114 | TraesCS1D02G016026 | 7381284 | 7384806 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_113 | TraesCS1D02G016100 | 7419157 | 7422949 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_9 | TraesCS1D02G016900 | 7592690 | 7609204 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_10 | TraesCS1D02G016983 | 7671168 | 7676063 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_11 | TraesCS1D02G016991 | 7678267 | 7680938 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_12 | TraesCS1D02G017400 | 7820918 | 7823449 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_13 | TraesCS1D02G017600 | 7868467 | 7872465 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_112 | TraesCS1D02G018700 | 8182627 | 8186066 | NB-LRR |
| Qstb-renan-1D | BST_expressed_pseudo_chr1D_nlr_111 | TraesCS1D02G018800 | 8226547 | 8230158 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_110 | TraesCS1D02G019600 | 8605145 | 8610344 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_14 | TraesCS1D02G019700 | 8610887 | 8616204 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_109 | TraesCS1D02G020619 | 8838803 | 8842102 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_108 | TraesCS1D02G021000 | 9028322 | 9037195 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_106 | TraesCS1D02G021200 | 9086119 | 9091764 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_104 | TraesCS1D02G021751 | 9309301 | 9328352 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_16 | TraesCS1D02G022500 | 9575753 | 9593333 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_102 | TraesCS1D02G026000 | 10661025 | 10664946 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_17 | TraesCS1D02G028200 | 11175841 | 11182532 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_18 | TraesCS1D02G028600 | 11272449 | 11278362 | NB-LRR |
| Qstb-renan-1D | BST_expressed_pseudo_chr1D_nlr_19 | TraesCS1D02G028700 | 11287245 | 11292876 | NB-LRR |
| Qstb-renan-1D | BST_pseudo_chr1D_nlr_20 | TraesCS1D02G028736 | 11319796 | 11321348 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_21 | TraesCS1D02G029000 | 11408761 | 11415088 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_22 | TraesCS1D02G029100 | 11451423 | 11459353 | NB-LRR |
| Qstb-renan-1D | BST_chr1D_nlr_23 | TraesCS1D02G029200 | 11493627 | 11499140 | NB-LRR |
| Qstb-renan-1D | TaWAK38_1D-gene | TraesCS1D02G016200 | 7429822 | 7445013 | WAK |
| Qstb-renan-1D | TaWAK39_1D-gene | TraesCS1D02G016800 | 7583590 | 7587977 | WAK |
| Qstb-renan-1D | TaWAK40_1D-gene | TraesCS1D02G017700 | 7874518 | 7876881 | WAK |
| Qstb-renan-1D | TaWAK41_1D-gene | TraesCS1D02G017800 | 7877418 | 7880329 | WAK |
| Qstb-renan-1D | TaWAK42_1D-gene | TraesCS1D02G017900 | 7896854 | 7899155 | WAK |
| Qstb-renan-5D | TaWAK349_5D-gene | TraesCS5D02G043400 | 42925913 | 42928461 | WAK |
| Qstb-renan-5D | TaWAK350_5D-gene | TraesCS5D02G043500 | 42930408 | 42932902 | WAK |
| Qstb-renan-5D | TaWAK351_5D-gene | TraesCS5D02G043532 | 42944893 | 42947212 | WAK |
| Qstb-renan-5D | TaWAK352_5D-gene | TraesCS5D02G052500 | 50569632 | 50576495 | WAK |
| Qstb-renan-5D | TaWAK353_5D-gene | TraesCS5D02G052800 | 50635242 | 50646756 | WAK |
| Qstb-renan-5D | TaWAK354_5D-gene | TraesCS5D02G061800 | 58138943 | 58142318 | WAK |
| Qstb-renan-5D | TaWAK355_5D-gene | TraesCS5D02G061900 | 58143379 | 58149665 | WAK |
| Qstb-renan-5D | TaWAK356_5D-gene | TraesCS5D02G062100 | 58151124 | 58155524 | WAK |
| Qstb-renan-5D | TaWAK357_5D-gene | TraesCS5D02G062200 | 58226914 | 58230221 | WAK |
| Qstb-renan-5D | TaWAK358_5D-gene | TraesCS5D02G062600 | 58419864 | 58422609 | WAK |
| Qstb-renan-5D | TaWAK359_5D-gene | TraesCS5D02G073900 | 72901902 | 72907097 | WAK |
| Qstb-renan-5D | TaWAK360_5D-gene | TraesCS5D02G096200 | 106519841 | 106525422 | WAK |
| Qstb-renan-7B | TaWAK556_7B-gene | TraesCS7B02G463200 | 720131495 | 720134235 | WAK |
| Qstb-renan-1D | TraesCS1D02G026200 | TraesCS1D02G026200 | 10715309 | 10722269 | Probable serine/threonine-protein kinase WNK3 |
| Qstb-renan-5D | TraesCS5D02G060900 | TraesCS5D02G060900 | 57843934 | 57851315 | Non-specific serine/threonine protein kinase |
| Qstb-renan-5D | TraesCS5D02G065700 | TraesCS5D02G065700 | 61052683 | 61060726 | Phosphatidylinositol 3-kinase VPS34 |
| Qstb-renan-5D | TraesCS5D02G068700 | TraesCS5D02G068700 | 65753632 | 65755248 | Non-specific serine/threonine protein kinase |
| Qstb-renan-5D | TraesCS5D02G069700 | TraesCS5D02G069700 | 67578001 | 67588803 | pfkB-like carbohydrate kinase family protein |
| Qstb-renan-5D | TraesCS5D02G081700 | TraesCS5D02G081700 | 82186877 | 82189457 | Serine/threonine protein kinase%2C Abscisic acid (ABA)-activated protein kinase%2C Hyperosmotic stress response%2C ABA signal transduction |
| Qstb-renan-5D | TraesCS5D02G089700 | TraesCS5D02G089700 | 97036711 | 97041067 | Diacylglycerol kinase |
| Qstb-renan-5D | TraesCS5D02G091000 | TraesCS5D02G091000 | 98227410 | 98230028 | L-type lectin-domain containing receptor kinase S.4 |
| Qstb-renan-5D | TraesCS5D02G104600 | TraesCS5D02G104600 | 118455172 | 118460088 | Nucleoside diphosphate kinase |
| Qstb-renan-5D | TraesCS5D02G104900 | TraesCS5D02G104900 | 118834967 | 118838504 | ATP-dependent 6-phosphofructokinase |
| Qstb-renan-5D | TraesCS5D02G120500 | TraesCS5D02G120500 | 170376901 | 170381844 | Diacylglycerol kinase |
| Qstb-renan-5D | TraesCS5D02G138800 | TraesCS5D02G138800 | 221007037 | 221012985 | Pyruvate kinase |
| Qstb-renan-5D | TraesCS5D02G140700 | TraesCS5D02G140700 | 224325320 | 224328774 | Phosphatidylinositol 4-phosphate 5-kinase |
| Qstb-renan-5D | TraesCS5D02G144800 | TraesCS5D02G144800 | 231350992 | 231353762 | Non-specific serine/threonine protein kinase |
| Qstb-renan-5D | TraesCS5D02G145100 | TraesCS5D02G145100 | 231743581 | 231750603 | Mitogen-activated protein kinase |
| Qstb-renan-5D | TraesCS5D02G166400 | TraesCS5D02G166400 | 259233864 | 259236422 | Receptor like protein kinase S.2 |
| Qstb-renan-5D | TraesCS5D02G181500 | TraesCS5D02G181500 | 282151742 | 282156543 | BR receptor kinase%2C Brassinosteroid (BR) perception in the roo |
| Qstb-renan-5D | TraesCS5D02G191900 | TraesCS5D02G191900 | 294637785 | 294639948 | NAD(H) kinase 3 |
| Qstb-renan-5D | TraesCS5D02G203600 | TraesCS5D02G203600 | 308863403 | 308865114 | Serine/threonine-protein kinase BLUS1 |
| Qstb-renan-5D | TraesCS5D02G214300 | TraesCS5D02G214300 | 323911872 | 323914256 | Serine/threonine-protein kinase |
| Qstb-renan-5D | TraesCS5D02G232500 | TraesCS5D02G232500 | 339652596 | 339654075 | Non-specific serine/threonine protein kinase |
| Qstb-renan-5D | TraesCS5D02G232600 | TraesCS5D02G232600 | 339712134 | 339713477 | Non-specific serine/threonine protein kinase |
| Qstb-renan-5D | TraesCS5D02G234000 | TraesCS5D02G234000 | 341192646 | 341202493 | ATP-dependent 6-phosphofructokinase |
| Qstb-renan-7B | TraesCS7B02G466300 | TraesCS7B02G466300 | 723900282 | 723903056 | Serine/threonine-protein kinase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Langlands-Perry, C.; Cuenin, M.; Bergez, C.; Krima, S.B.; Gélisse, S.; Sourdille, P.; Valade, R.; Marcel, T.C. Resistance of the Wheat Cultivar ‘Renan’ to Septoria Leaf Blotch Explained by a Combination of Strain Specific and Strain Non-Specific QTL Mapped on an Ultra-Dense Genetic Map. Genes 2022, 13, 100. https://doi.org/10.3390/genes13010100
Langlands-Perry C, Cuenin M, Bergez C, Krima SB, Gélisse S, Sourdille P, Valade R, Marcel TC. Resistance of the Wheat Cultivar ‘Renan’ to Septoria Leaf Blotch Explained by a Combination of Strain Specific and Strain Non-Specific QTL Mapped on an Ultra-Dense Genetic Map. Genes. 2022; 13(1):100. https://doi.org/10.3390/genes13010100
Chicago/Turabian StyleLanglands-Perry, Camilla, Murielle Cuenin, Christophe Bergez, Safa Ben Krima, Sandrine Gélisse, Pierre Sourdille, Romain Valade, and Thierry C. Marcel. 2022. "Resistance of the Wheat Cultivar ‘Renan’ to Septoria Leaf Blotch Explained by a Combination of Strain Specific and Strain Non-Specific QTL Mapped on an Ultra-Dense Genetic Map" Genes 13, no. 1: 100. https://doi.org/10.3390/genes13010100
APA StyleLanglands-Perry, C., Cuenin, M., Bergez, C., Krima, S. B., Gélisse, S., Sourdille, P., Valade, R., & Marcel, T. C. (2022). Resistance of the Wheat Cultivar ‘Renan’ to Septoria Leaf Blotch Explained by a Combination of Strain Specific and Strain Non-Specific QTL Mapped on an Ultra-Dense Genetic Map. Genes, 13(1), 100. https://doi.org/10.3390/genes13010100