
Thirteen Independent Genetic Loci Associated with Preserved Processing Speed in a Study of Cognitive Resilience in 330,097 Individuals in the UK Biobank


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. The UK Biobank
2.3. Genetic Data
2.4. Phenotypic Data
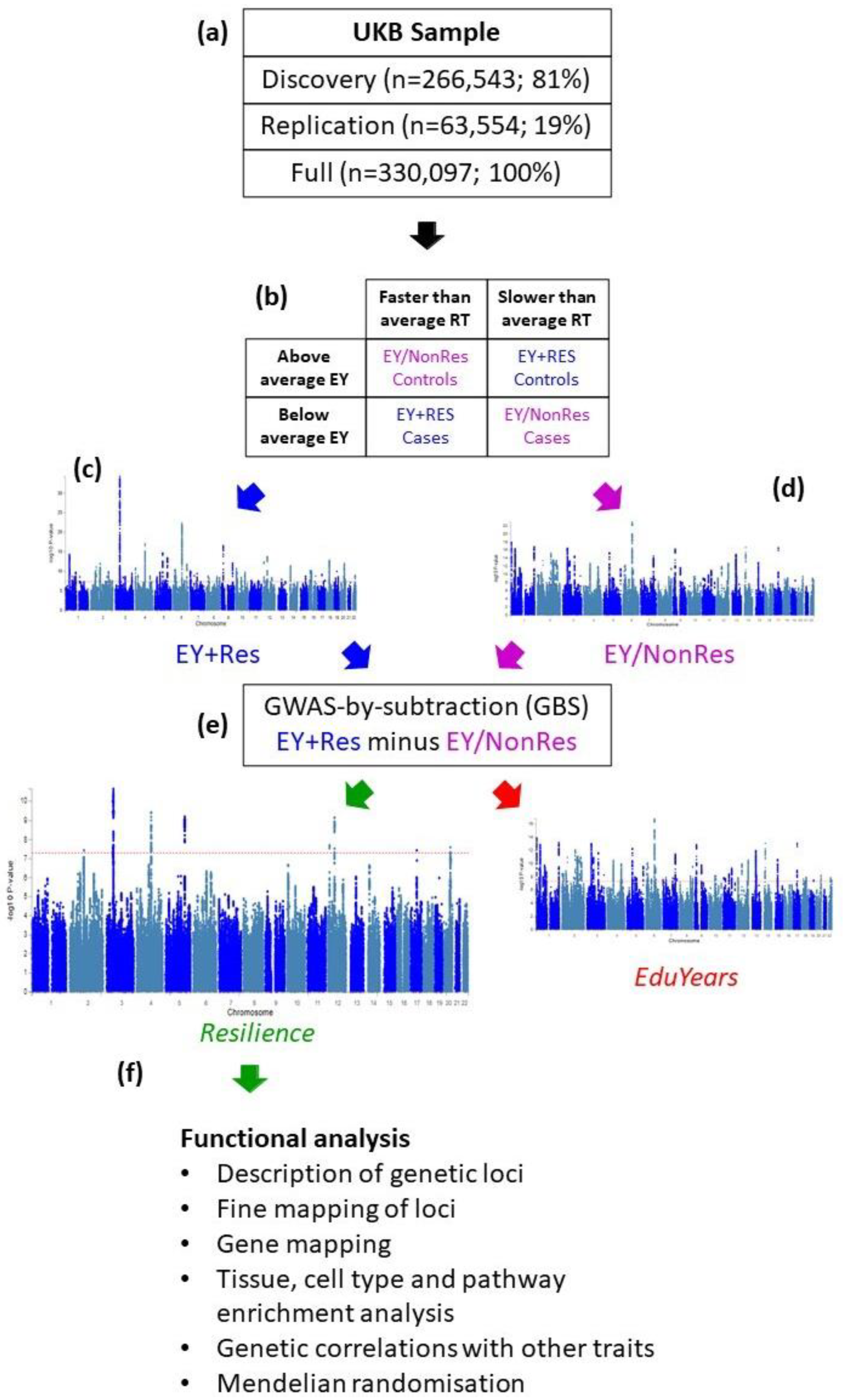
2.5. Generation of Resilience Phenotype
2.6. GWAS-by-Subtraction (GBS)
2.7. Execution of GBS
2.8. Calculation of Sample Size after GBS
2.9. Additional GWAS
2.10. Identification of Genomic Loci Associated with Resilience
2.11. Function Analysis of GWAS Output
2.12. Mapping SNPs to Genes
2.13. MAGMA Gene-Based Analysis
2.14. Comparison with Published Traits
2.15. Mendelian Randomisation
2.16. US Health and Retirement Study (HRS)
2.17. Genetic Data
2.18. Cognitive Phenotypes
2.19. GWAS of Cognitive Change over Time
3. Results
3.1. Initial Phenotype Development
3.2. Discovery and Replication Analysis within the UKB
3.3. Analysis of the Full Sample
3.4. Analysis in a Dataset Independent of the UKB
3.5. Functional Analysis
3.5.1. Description of Genetic Loci
3.5.2. Fine Mapping
3.5.3. Gene Mapping
3.5.4. Tissue, Cell Type and Pathway Enrichment Analysis
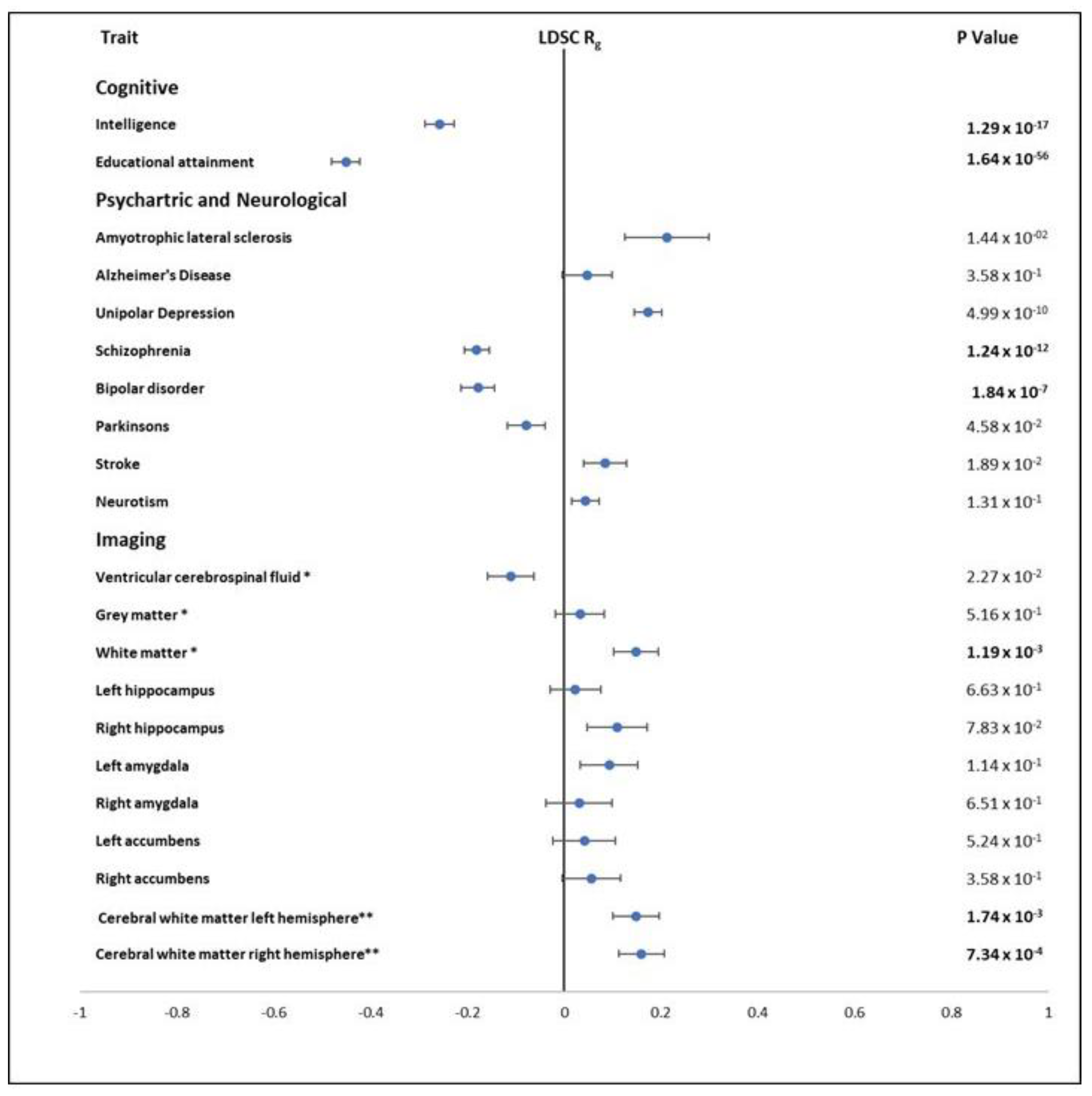
3.5.5. Genetic Correlations with Other Traits
3.5.6. Mendelian Randomisation
3.5.7. Examination of the Relationship of Resilience with RT
3.5.8. Effect of Large Locus on Chromosome 3
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
- UK Biobank: http://biobank.ndph.ox.ac.uk, accessed on 18 December 2021.
- Plink: www.cog-genomics.org/plink/2.0/, accessed on 18 December 2021.
- GenomicSEM: https://github.com/MichelNivard/GenomicSEM/wiki, accessed on 18 December 2021.
- GWAS-by-subtraction: https://rpubs.com/MichelNivard/565885, accessed on 18 December 2021.
- GBS sample size (N effective) calculation: https://github.com/PerlineDemange/non-cognitive/blob/master/GenomicSEM/Cholesky%20model/Calculation_samplesize.R, accessed on 18 December 2021
- Functional Mapping and Annotation (FUMA): https://fuma.ctglab.nl/downpage.html, accessed on 18 December 2021.
- Venn diagram: http://bioinformatics.psb.ugent.be/webtools/Venn/, accessed on 18 December 2021.
- FINEMAP: http://www.christianbenner.com/, accessed 18 December 2021.
- Generalised Summary-data-based MR: https://cnsgenomics.com/software/gcta/#GSMR, accessed on 18 December 2021.
- GWAS Catalog: https://www.ebi.ac.uk/gwas/, accessed on 18 December 2021.
- Brain phenotypes: https://open.win.ox.ac.uk/ukbiobank/big40/, accessed 18 December 2021.
- GWAS Atlas: https://atlas.ctglab.nl/, accessed 18 December 2021.
- Ensembl Variant Effect Predictor: https://www.ensembl.org/Tools/VEP, accessed on 18 December 2021
- HRS: https://hrsonline.isr.umich.edu/, accessed 18 December 2021
References
- Deary, I.J.; Corley, J.; Gow, A.J.; Harris, S.E.; Houlihan, L.M.; Marioni, R.E.; Penke, L.; Rafnsson, S.B.; Starr, J.M. Age-associated cognitive decline. Br. Med. Bull. 2009, 92, 135–152. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S.J.; Das, D.; Cherbuin, N.; Anstey, K.J.; Easteal, S. Association of genetic risk factors with cognitive decline: The PATH through life project. Neurobiol Aging 2016, 41, 150–158. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. World Report on Ageing and Health; World Health Organization: Geneva, Switzerland, 2015.
- Staal, M.A.; Bolton, A.; Yaroush, R.; Bourne, L., Jr. Cognitive Performance and Resilience to Stress; Routledge: London, UK, 2008; pp. 259–299. [Google Scholar]
- Cabeza, R.; Albert, M.; Belleville, S.; Craik, F.I.M.; Duarte, A.; Grady, C.L.; Lindenberger, U.; Nyberg, L.; Park, D.C.; Reuter-Lorenz, P.A.; et al. Maintenance, reserve and compensation: The cognitive neuroscience of healthy ageing. Nat. Rev. Neurosci. 2018, 19, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Stern, Y.; Arenaza-Urquijo, E.M.; Bartrés-Faz, D.; Belleville, S.; Cantilon, M.; Chetelat, G.; Ewers, M.; Franzmeier, N.; Kempermann, G.; Kremen, W.S.; et al. Whitepaper: Defining and investigating cognitive reserve, brain reserve, and brain maintenance. Alzheimer’s Dement. 2018, 16, 1305–1311. [Google Scholar] [CrossRef]
- Cadar, D.; Robitaille, A.; Pattie, A.; Deary, I.; Muniz-Terrera, G. The Long Arm of Childhood Intelligence on Terminal Decline: Evidence From the Lothian Birth Cohort 1921. Psychol Aging 2020, 35, 806–817. [Google Scholar] [CrossRef]
- Guerra-Carrillo, B.; Katovich, K.; Bunge, S.A. Does higher education hone cognitive functioning and learning efficacy? Findings from a large and diverse sample. PLoS ONE 2017, 12, e0182276. [Google Scholar] [CrossRef] [Green Version]
- Lövdén, M.; Fratiglioni, L.; Glymour, M.M.; Lindenberger, U.; Tucker-Drob, E.M. Education and Cognitive Functioning Across the Life Span. Psychol. Sci. Public Interest 2020, 21, 6–41. [Google Scholar] [CrossRef]
- Geary, D.C. The Spark of Life and the Unification of Intelligence, Health, and Aging. Curr. Dir. Psychol. Sci. 2019, 28, 223–228. [Google Scholar] [CrossRef]
- Salthouse, T. The processing-speed theory of adult age differences in cognition. Psychol. Rev. 1996, 103, 403–428. [Google Scholar] [CrossRef] [Green Version]
- Salthouse, T.A.; Ferrer-Caja, E. What needs to be explained to account for age-related effects on multiple cognitive variables? Psychol. Aging 2003, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Schubert, A.-L.; Nunez, M.D.; Hagemann, D.; Vandekerckhove, J. Individual Differences in Cortical Processing Speed Predict Cognitive Abilities: A Model-Based Cognitive Neuroscience Account. Comput. Brain Behav. 2019, 2, 64–84. [Google Scholar] [CrossRef] [Green Version]
- Scheiber, C.; Chen, H.; Kaufman, A.S.; Weiss, L.G. How Much Does WAIS-IV Perceptual Reasoning Decline Across the 20 to 90-Year Lifespan When Processing Speed is Controlled? Appl. Neuropsychol. Adult 2017, 24, 116–131. [Google Scholar] [CrossRef]
- Sudlow, C.; Gallacher, J.; Allen, N.; Beral, V.; Burton, P.; Danesh, J.; Downey, P.; Elliott, P.; Green, J.; Landray, M.; et al. UK biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015, 12, e1001779. [Google Scholar] [CrossRef] [Green Version]
- Deary, I.J. Intelligence. Curr. Biol. 2013, 23, R673–R676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, G.; Marioni, R.E.; Liewald, D.C.; Hill, W.D.; Hagenaars, S.P.; Harris, S.E.; Ritchie, S.J.; Luciano, M.; Fawns-Ritchie, C.; Lyall, D.; et al. Genome-wide association study of cognitive functions and educational attainment in UK Biobank (N=112,151). Mol. Psychiatry 2016, 21, 758–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plomin, R.; von Stumm, S. The new genetics of intelligence. Nat. Rev. Genet. 2018, 19, 148. [Google Scholar] [CrossRef] [PubMed]
- Rietveld, C.A.; Esko, T.; Davies, G.; Pers, T.H.; Turley, P.; Benyamin, B.; Chabris, C.F.; Emilsson, V.; Johnson, A.D.; Lee, J.J.; et al. Common genetic variants associated with cognitive performance identified using the proxy-phenotype method. Proc. Natl. Acad. Sci. USA 2014, 111, 13790–13794. [Google Scholar] [CrossRef] [Green Version]
- Gordon, R.; Smith-Spark, J.H.; Newton, E.J.; Henry, L.A. Executive Function and Academic Achievement in Primary School Children: The Use of Task-Related Processing Speed. Front. Psychol. 2018, 9, 582. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.J.; Wedow, R.; Okbay, A.; Kong, E.; Maghzian, O.; Zacher, M.; Nguyen-Viet, T.A.; Bowers, P.; Sidorenko, J.; Karlsson Linnér, R.; et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018, 50, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grotzinger, A.D.; Rhemtulla, M.; de Vlaming, R.; Ritchie, S.J.; Mallard, T.T.; David, H.W.; Ip, H.F.; Marioni, R.E.; McIntosh, A.M.; Deary, I.J.; et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat. Hum. Behav. 2019, 3, 513–525. [Google Scholar] [CrossRef]
- Demange, P.A.; Malanchini, M.; Mallard, T.T.; Biroli, P.; Cox, S.R.; Grotzinger, A.D.; Tucker-Drob, E.M.; Abdellaoui, A.; Arseneault, L.; van Bergen, E.; et al. Investigating the genetic architecture of noncognitive skills using GWAS-by-subtraction. Nat. Genet. 2021, 53, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fawns-Ritchie, C.; Deary, I.J. Reliability and validity of the UK Biobank cognitive tests. PLoS ONE 2020, 15, e0231627. [Google Scholar] [CrossRef]
- Lyall, D.M.; Cullen, B.; Allerhand, M.; Smith, D.J.; Mackay, D.; Evans, J.; Anderson, J.; Fawns-Ritchie, C.; McIntosh, A.M.; Deary, I.J.; et al. Cognitive Test Scores in UK Biobank: Data Reduction in 480,416 Participants and Longitudinal Stability in 20,346 Participants. PLoS ONE 2016, 11, e0154222. [Google Scholar] [CrossRef] [Green Version]
- IBM Corp. IBM SPSS Statistics for Windows; Released 2016; IBM Corp: Armonk, NY, USA, 2016. [Google Scholar]
- Nivard, M. GenomicSem. Available online: https://github.com/MichelNivard/GenomicSEM/wiki (accessed on 18 December 2021).
- Chang, C.C.; Chow, C.C.; Tellier, L.C.A.M.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 2015, 4, s13742–015. [Google Scholar] [CrossRef] [PubMed]
- Mallard, T.T.; Linnér, R.K.; Grotzinger, A.D.; Sanchez-Roige, S.; Seidlitz, J.; Okbay, A.; de Vlaming, R.; Meddens, S.F.W.; Palmer, A.A.; Davis, L.K.; et al. Multivariate GWAS of psychiatric disorders and their cardinal symptoms reveal two dimensions of cross-cutting genetic liabilities. BioRxiv 2020, 603134. [Google Scholar] [CrossRef]
- Watanabe, K.; Taskesen, E.; van Bochoven, A.; Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 2017, 8, 1826. [Google Scholar] [CrossRef] [Green Version]
- Benner, C.; Spencer, C.C.A.; Havulinna, A.S.; Salomaa, V.; Ripatti, S.; Pirinen, M. FINEMAP: Efficient variable selection using summary data from genome-wide association studies. Bioinformatics 2016, 32, 1493–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, I.E.; Savage, J.E.; Watanabe, K.; Bryois, J.; Williams, D.M.; Steinberg, S.; Sealock, J.; Karlsson, I.K.; Hägg, S.; Athanasiu, L.; et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat. Genet. 2019, 51, 404–413. [Google Scholar] [CrossRef]
- Savage, J.E.; Jansen, P.R.; Stringer, S.; Watanabe, K.; Bryois, J.; de Leeuw, C.A.; Nagel, M.; Awasthi, S.; Barr, P.B.; Coleman, J.R.I.; et al. Genome-wide association meta-analysis in 269,867 individuals identifies new genetic and functional links to intelligence. Nat. Genet. 2018, 50, 912–919. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.P.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362, eaat8464. [Google Scholar] [CrossRef] [Green Version]
- Bonder, M.J.; Luijk, R.; Zhernakova, D.V.; Moed, M.; Deelen, P.; Vermaat, M.; van Iterson, M.; van Dijk, F.; van Galen, M.; Bot, J.; et al. Disease variants alter transcription factor levels and methylation of their binding sites. Nat. Genet. 2017, 49, 131–138. [Google Scholar] [CrossRef] [Green Version]
- Westra, H.-J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic identification of trans eQTLs as putative drivers of known disease associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battle, A.; Brown, C.D.; Engelhardt, B.E.; Montgomery, S.B. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreservation Biobanking 2015, 13, 311–319. [CrossRef] [PubMed] [Green Version]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015, 1, 417–425. [Google Scholar] [CrossRef] [Green Version]
- Buniello, A.; MacArthur, J.A.L.; Cerezo, M.; Harris, L.W.; Hayhurst, J.; Malangone, C.; McMahon, A.; Morales, J.; Mountjoy, E.; Sollis, E.; et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 2019, 47, D1005–D1012. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, K.; Umićević Mirkov, M.; de Leeuw, C.A.; van den Heuvel, M.P.; Posthuma, D. Genetic mapping of cell type specificity for complex traits. Nat. Commun. 2019, 10, 3222. [Google Scholar] [CrossRef] [Green Version]
- Zhong, S.; Zhang, S.; Fan, X.; Wu, Q.; Yan, L.; Dong, J.; Zhang, H.; Li, L.; Sun, L.; Pan, N.; et al. A single-cell RNA-seq survey of the developmental landscape of the human prefrontal cortex. Nature 2018, 555, 524–528. [Google Scholar] [CrossRef]
- Darmanis, S.; Sloan, S.A.; Zhang, Y.; Enge, M.; Caneda, C.; Shuer, L.M.; Hayden Gephart, M.G.; Barres, B.A.; Quake, S.R. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl. Acad. Sci. USA 2015, 112, 7285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeisel, A.; Hochgerner, H.; Lönnerberg, P.; Johnsson, A.; Memic, F.; van der Zwan, J.; Häring, M.; Braun, E.; Borm, L.E.; La Manno, G.; et al. Molecular Architecture of the Mouse Nervous System. Cell 2018, 174, 999–1014.e1022. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Zheng, Z.; Zhang, F.; Wu, Y.; Trzaskowski, M.; Maier, R.; Robinson, M.R.; McGrath, J.J.; Visscher, P.M.; Wray, N.R.; et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 2018, 9, 224. [Google Scholar] [CrossRef] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [Green Version]
- Field, A. Discovering Statistics Using IBM SPSS Statistics; Sage: New York, NY, USA, 2013. [Google Scholar]
- Faul, J.; Smith, J.; Zhao, W. Health and Retirement Study: Candidate Gene and SNP Data Description. Health Retire. Study, Univ. Mich., Ann. Arbor, MI. Google Scholar Article Location. 2014. Available online: https://hrs.isr.umich.edu/sites/default/files/genetic/candidategene/CandidateGeneSNPDataDescription.pdf (accessed on 28 December 2021).
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, G.; Lam, M.; Harris, S.E.; Trampush, J.W.; Luciano, M.; Hill, W.D.; Hagenaars, S.P.; Ritchie, S.J.; Marioni, R.E.; Fawns-Ritchie, C.; et al. Study of 300,486 individuals identifies 148 independent genetic loci influencing general cognitive function. Nat. Commun. 2018, 9, 2098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasaki, S.; Gaiteri, C.; Mostafavi, S.; Yu, L.; Wang, Y.; De Jager, P.L.; Bennett, D.A. Multi-omic Directed Networks Describe Features of Gene Regulation in Aged Brains and Expand the Set of Genes Driving Cognitive Decline. Front. Genet. 2018, 9, 294. [Google Scholar] [CrossRef]
- Annamneedi, A.; Caliskan, G.; Müller, S.; Montag, D.; Budinger, E.; Angenstein, F.; Fejtova, A.; Tischmeyer, W.; Gundelfinger, E.D.; Stork, O. Ablation of the presynaptic organizer Bassoon in excitatory neurons retards dentate gyrus maturation and enhances learning performance. Brain Struct. Funct. 2018, 223, 3423–3445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.-G.; Zhang, S.; Park, H.; Park, S.J.; Kim, S.; Chung, C. Inositol hexakisphosphate kinase-1 is a key mediator of prepulse inhibition and short-term fear memory. Mol. Brain 2020, 13, 72. [Google Scholar] [CrossRef]
- Wang, P.; Geng, J.; Gao, J.; Zhao, H.; Li, J.; Shi, Y.; Yang, B.; Xiao, C.; Linghu, Y.; Sun, X.; et al. Macrophage achieves self-protection against oxidative stress-induced ageing through the Mst-Nrf2 axis. Nat. Commun. 2019, 10, 755. [Google Scholar] [CrossRef] [PubMed]
- Gontier, G.; Iyer, M.; Shea, J.M.; Bieri, G.; Wheatley, E.G.; Ramalho-Santos, M.; Villeda, S.A. Tet2 Rescues Age-Related Regenerative Decline and Enhances Cognitive Function in the Adult Mouse Brain. Cell Rep. 2018, 22, 1974–1981. [Google Scholar] [CrossRef] [Green Version]
- Gardiner, S.L.; Trompet, S.; Sabayan, B.; Boogaard, M.W.; Jukema, J.W.; Slagboom, P.E.; Roos, R.A.C.; van der Grond, J.; Aziz, N.A. Repeat variations in polyglutamine disease–associated genes and cognitive function in old age. Neurobiol. Aging 2019, 84, 236.e217–236.e228. [Google Scholar] [CrossRef]
- Lastres-Becker, I.; Nonis, D.; Nowock, J.; Auburger, G. New alternative splicing variants of the ATXN2 transcript. Neurol. Res. Pract. 2019, 1, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmers, P.R.; Mounier, N.; Lall, K.; Fischer, K.; Ning, Z.; Feng, X.; Bretherick, A.D.; Clark, D.W.; eQTLGen Consortium; Agbessi, M.; et al. Genomics of 1 million parent lifespans implicates novel pathways and common diseases and distinguishes survival chances. Elife 2019, 8, e39856. [Google Scholar] [CrossRef] [PubMed]
- Melzer, D.; Pilling, L.C.; Ferrucci, L. The genetics of human ageing. Nat. Rev. Genet. 2020, 21, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.N.; Ren, J. Aldehyde Dehydrogenase 2 (ALDH2) and Aging: Is There a Sensible Link? Adv. Exp. Med. Biol. 2019, 1193, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.C.; Kotermanski, S.E.; Kochanek, P.M. Whole-transcriptome microarray analysis reveals regulation of Rab4 by RBM5 in neurons. Neuroscience 2017, 361, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Panzanelli, P.; Früh, S.; Fritschy, J.-M. Differential role of GABAA receptors and neuroligin 2 for perisomatic GABAergic synapse formation in the hippocampus. Brain Struct. Funct. 2017, 222, 4149–4161. [Google Scholar] [CrossRef] [Green Version]
- McLaren, W.; Gil, L.; Hunt, S.E.; Riat, H.S.; Ritchie, G.R.S.; Thormann, A.; Flicek, P.; Cunningham, F. The Ensembl Variant Effect Predictor. Genome Biol. 2016, 17, 122. [Google Scholar] [CrossRef] [Green Version]
- The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648. [CrossRef] [Green Version]
- Rozycka, A.; Liguz-Lecznar, M. The space where aging acts: Focus on the GABAergic synapse. Aging Cell 2017, 16, 634–643. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- de Leeuw, C.A.; Mooij, J.M.; Heskes, T.; Posthuma, D. MAGMA: Generalized Gene-Set Analysis of GWAS Data. PLOS Comput. Biol. 2015, 11, e1004219. [Google Scholar] [CrossRef] [PubMed]
- Palomer, E.; Buechler, J.; Salinas, P.C. Wnt Signaling Deregulation in the Aging and Alzheimer’s Brain. Front. Cell. Neurosci. 2019, 13, 227. [Google Scholar] [CrossRef]
- Nagel, M.; Jansen, P.R.; Stringer, S.; Watanabe, K.; de Leeuw, C.A.; Bryois, J.; Savage, J.E.; Hammerschlag, A.R.; Skene, N.G.; Muñoz-Manchado, A.B.; et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018, 50, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pardiñas, A.F.; Holmans, P.; Pocklington, A.J.; Escott-Price, V.; Ripke, S.; Carrera, N.; Legge, S.E.; Bishop, S.; Cameron, D.; Hamshere, M.L.; et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat. Genet. 2018, 50, 381–389. [Google Scholar] [CrossRef] [Green Version]
- Stahl, E.A.; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.I.; Gaspar, H.A.; et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019, 51, 793–803. [Google Scholar] [CrossRef] [PubMed]
- Luciano, M.; Hagenaars, S.P.; Davies, G.; Hill, W.D.; Clarke, T.-K.; Shirali, M.; Harris, S.E.; Marioni, R.E.; Liewald, D.C.; Fawns-Ritchie, C.; et al. Association analysis in over 329,000 individuals identifies 116 independent variants influencing neuroticism. Nat. Genet. 2018, 50, 6–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Rheenen, W.; Shatunov, A.; Dekker, A.M.; McLaughlin, R.L.; Diekstra, F.P.; Pulit, S.L.; van der Spek, R.A.A.; Võsa, U.; de Jong, S.; Robinson, M.R.; et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat. Genet. 2016, 48, 1043–1048. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Blauwendraat, C.; Vallerga, C.L.; Heilbron, K.; Bandres-Ciga, S.; Chang, D.; Tan, M.; Kia, D.A.; Noyce, A.J.; Xue, A.; et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 2019, 18, 1091–1102. [Google Scholar] [CrossRef]
- Toepper, M. Dissociating Normal Aging from Alzheimer’s Disease: A View from Cognitive Neuroscience. J. Alzheimers Dis. 2017, 57, 331–352. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Douaud, G.; Chen, W.; Hanayik, T.; Alfaro-Almagro, F.; Sharp, K.; Elliott, L.T. Enhanced Brain Imaging Genetics in UK Biobank. BioRxiv 2020, 2020.07.27.223545. [Google Scholar] [CrossRef]
- Nave, G.; Jung, W.H.; Karlsson Linnér, R.; Kable, J.W.; Koellinger, P.D. Are Bigger Brains Smarter? Evidence From a Large-Scale Preregistered Study. Psychol. Sci. 2019, 30, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Orellana, C.; Ferreira, D.; Muehlboeck, J.S.; Mecocci, P.; Vellas, B.; Tsolaki, M.; Kłoszewska, I.; Soininen, H.; Lovestone, S.; Simmons, A.; et al. Measuring Global Brain Atrophy with the Brain Volume/Cerebrospinal Fluid Index: Normative Values, Cut-Offs and Clinical Associations. Neuro Degener. Dis. 2016, 16, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Lisman, J.; Buzsáki, G.; Eichenbaum, H.; Nadel, L.; Ranganath, C.; Redish, A.D. Viewpoints: How the hippocampus contributes to memory, navigation and cognition. Nat. Neurosci. 2017, 20, 1434–1447. [Google Scholar] [CrossRef]
- Floresco, S.B. The nucleus accumbens: An interface between cognition, emotion, and action. Annu. Rev. Psychol. 2015, 66, 25–52. [Google Scholar] [CrossRef] [PubMed]
- Henry, A.; Katsoulis, M.; Masi, S.; Fatemifar, G.; Denaxas, S.; Acosta, D.; Garfield, V.; Dale, C.E. The relationship between sleep duration, cognition and dementia: A Mendelian randomization study. Int. J. Epidemiol. 2019, 48, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Low, D.V.; Wu, M.N.; Spira, A.P. Sleep Duration and Cognition in a Nationally Representative Sample of U.S. Older Adults. Am. J. Geriatr. Psychiatry 2019, 27, 1386–1396. [Google Scholar] [CrossRef]
- Coleman, J.R.I.; Bryois, J.; Gaspar, H.A.; Jansen, P.R.; Savage, J.E.; Skene, N.; Plomin, R.; Muñoz-Manchado, A.B.; Linnarsson, S.; Crawford, G.; et al. Biological annotation of genetic loci associated with intelligence in a meta-analysis of 87,740 individuals. Mol. Psychiatry 2019, 24, 182–197. [Google Scholar] [CrossRef] [Green Version]
- Eckert, M.A. Slowing Down: Age-Related Neurobiological Predictors of Processing Speed. Front. Neurosci. 2011, 5, 25. [Google Scholar] [CrossRef] [Green Version]
- Bott, N.T.; Bettcher, B.M.; Yokoyama, J.S.; Frazier, D.T.; Wynn, M.; Karydas, A.; Yaffe, K.; Kramer, J.H. Youthful Processing Speed in Older Adults: Genetic, Biological, and Behavioral Predictors of Cognitive Processing Speed Trajectories in Aging. Front. Aging Neurosci. 2017, 9, 55. [Google Scholar] [CrossRef] [Green Version]
- Seblova, D.; Berggren, R.; Lövdén, M. Education and age-related decline in cognitive performance: Systematic review and meta-analysis of longitudinal cohort studies. Ageing Res. Rev. 2020, 58, 101005. [Google Scholar] [CrossRef] [PubMed]
- Tikhomirova, T.; Malykh, A.; Malykh, S. Predicting Academic Achievement with Cognitive Abilities: Cross-Sectional Study across School Education. Behav. Sci. 2020, 10, 158. [Google Scholar] [CrossRef] [PubMed]
- Opdebeeck, C.; Martyr, A.; Clare, L. Cognitive reserve and cognitive function in healthy older people: A meta-analysis. Aging Neuropsychol. Cogn. 2016, 23, 40–60. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzgerald, J.; Fahey, L.; Holleran, L.; Ó Broin, P.; Donohoe, G.; Morris, D.W. Thirteen Independent Genetic Loci Associated with Preserved Processing Speed in a Study of Cognitive Resilience in 330,097 Individuals in the UK Biobank. Genes 2022, 13, 122. https://doi.org/10.3390/genes13010122
Fitzgerald J, Fahey L, Holleran L, Ó Broin P, Donohoe G, Morris DW. Thirteen Independent Genetic Loci Associated with Preserved Processing Speed in a Study of Cognitive Resilience in 330,097 Individuals in the UK Biobank. Genes. 2022; 13(1):122. https://doi.org/10.3390/genes13010122
Chicago/Turabian StyleFitzgerald, Joan, Laura Fahey, Laurena Holleran, Pilib Ó Broin, Gary Donohoe, and Derek W. Morris. 2022. "Thirteen Independent Genetic Loci Associated with Preserved Processing Speed in a Study of Cognitive Resilience in 330,097 Individuals in the UK Biobank" Genes 13, no. 1: 122. https://doi.org/10.3390/genes13010122