
Genome Sequence of Chrysotila roscoffensis, a Coccolithphore Contributed to Global Biogeochemical Cycles

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. C. roscoffensis Strain and DNA Extraction
2.2. Library Construction and Sequencing
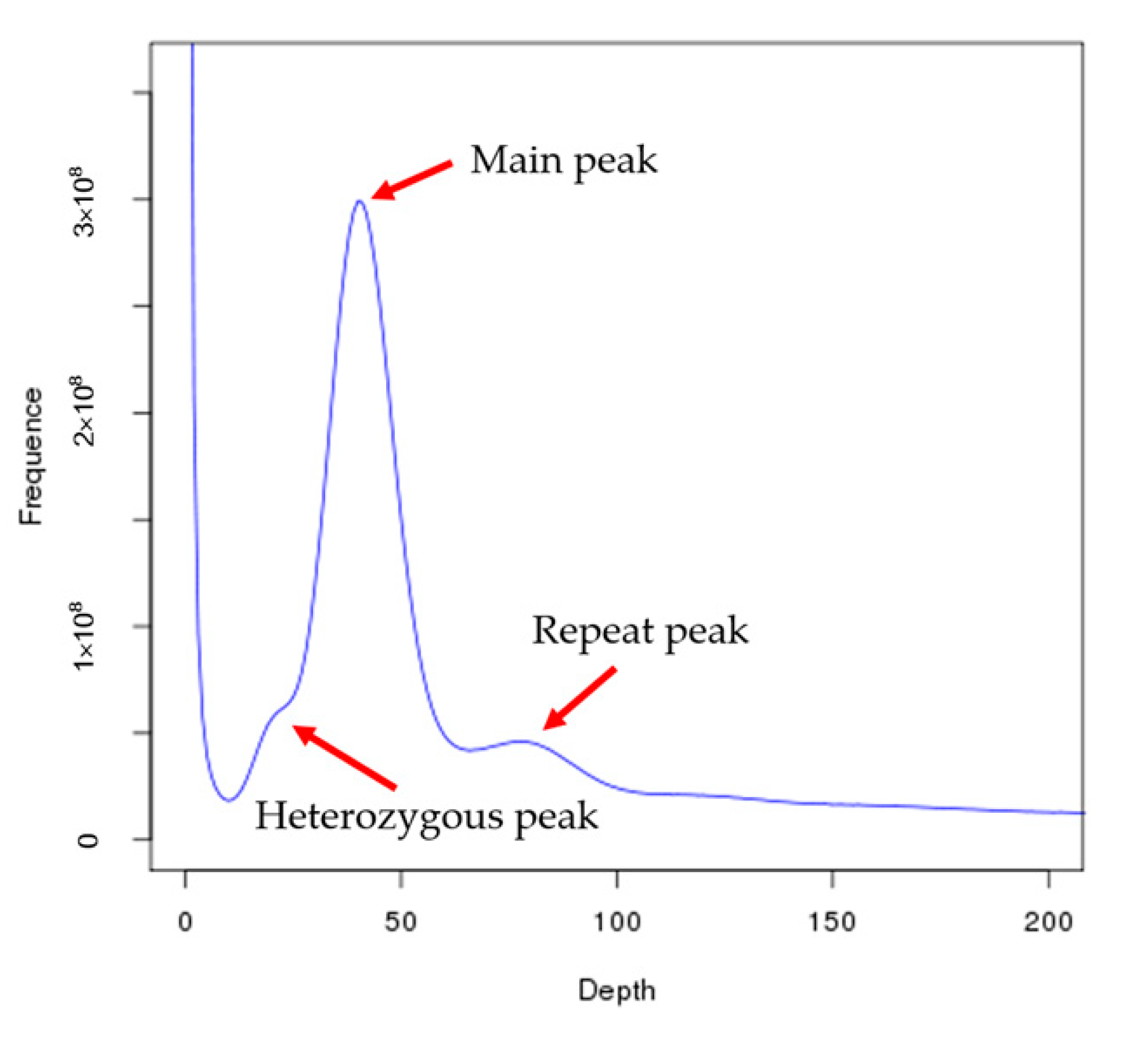
2.3. Genome Size Estimation and De Novo Genome Assembly
2.4. Repetitive Sequences Annotation
2.5. Genome Annotation
2.6. Phylogenetic and Comparative Genomic Analysis
3. Results and Discussion
3.1. Genome Analysis of C. roscoffensis
3.2. Genome Annotation
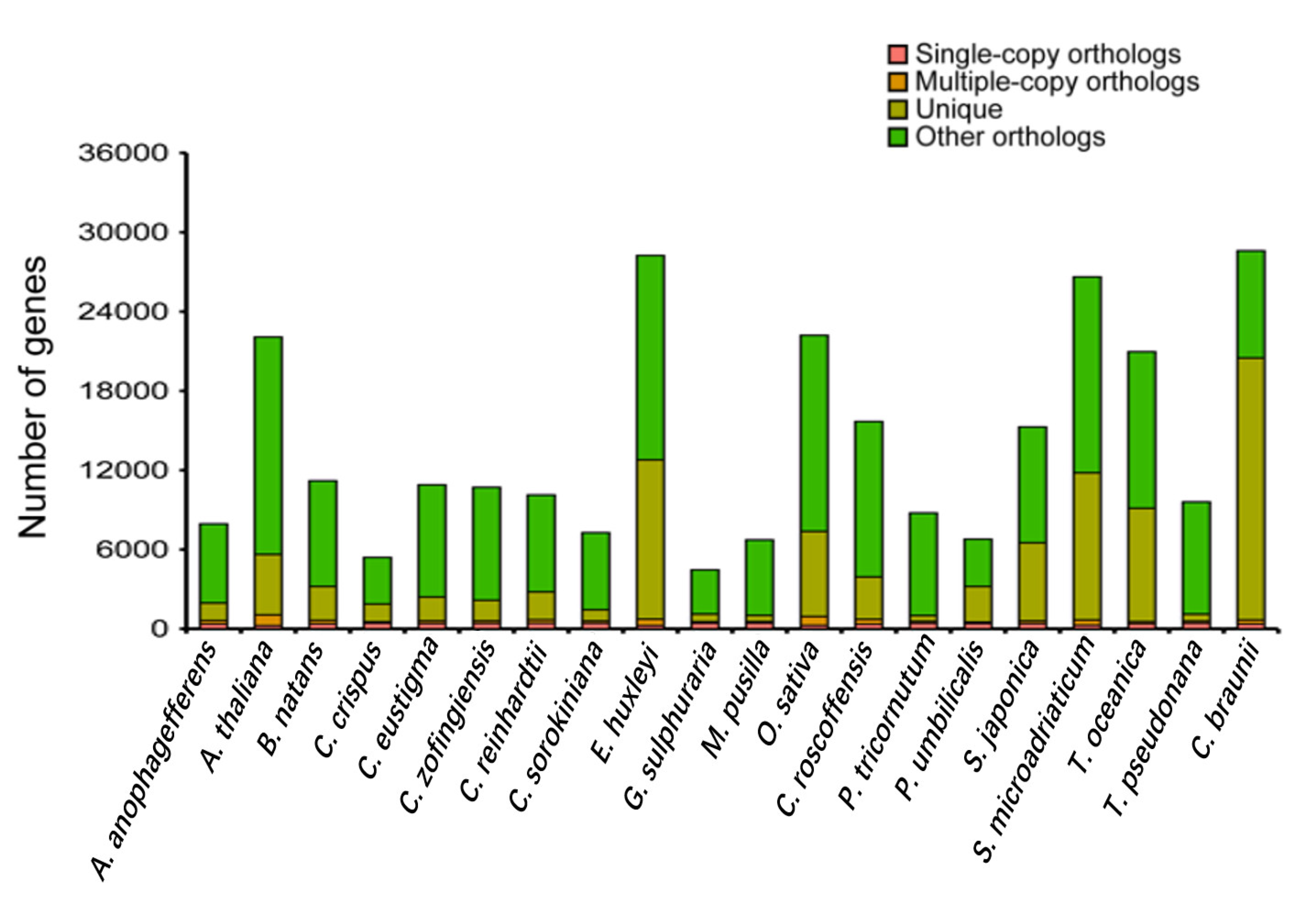
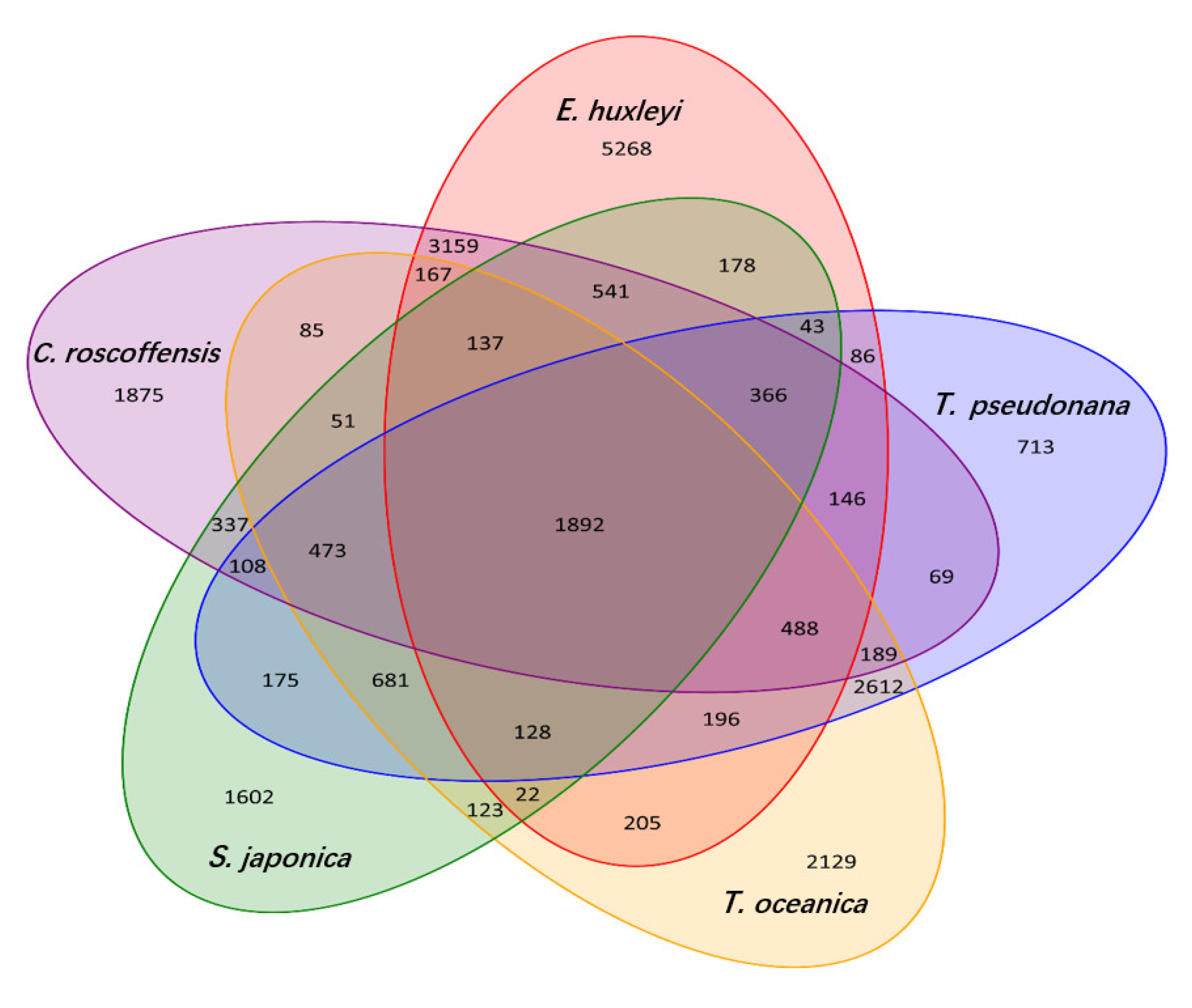
3.3. Phylogenetic and Comparative Genomic Analysis
3.4. Expanded Coccoliths-Related Gene Families
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Holligan, P.M.; Fernández, E.; Aiken, J.; Balch, W.M.; Boyd, P.; Burkill, P.H.; Finch, M.; Groom, S.B.; Malin, G.; Muller, K.; et al. A biogeochemical study of the coccolithophore, Emiliania huxleyi, in the North Atlantic. Glob. Biogeochem. Cycles 1993, 7, 879–900. [Google Scholar] [CrossRef]
- Rost, B.; Riebesell, U. Coccolithophores and the biological pump: Responses to environmental changes. Coccolithophores 2004, 99–125. [Google Scholar] [CrossRef] [Green Version]
- Poulton, A.J.; Adey, T.R.; Balch, W.M.; Holligan, P.M. Relating coccolithophore calcification rates to phytoplankton community dynamics: Regional differences and implications for carbon export. Deep. Sea Res. Part II Top. Stud. Oceanogr. 2007, 54, 538–557. [Google Scholar] [CrossRef]
- Milliman, J.D. Production and accumulation of calcium carbonate in the ocean: Budget of a nonsteady state. Glob. Biogeochem. Cycles 1993, 7, 927–957. [Google Scholar] [CrossRef]
- Taylor, A.R.; Brownlee, C.; Wheeler, G. Coccolithophore Cell Biology: Chalking Up Progress. Annu. Rev. Mar. Sci. 2017, 9, 283–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaas, C.; Archer, D.E. Association of sinking organic matter with various types of mineral ballast in the deep sea: Implications for the rain ratio. Glob. Biogeochem. Cycles 2002, 16, 63-1–63-14. [Google Scholar] [CrossRef]
- Van der Wal, P.; de Jong, E.; Westbroek, P.; de Bruijn, W.; Mulder-Stapel, A. Polysaccharide localization, coccolith formation, and golgi dynamics in the coccolithophorid Hymenomonas carterae. J. Ultrastruct. Res. 1983, 85, 139–158. [Google Scholar] [CrossRef]
- Marsh, M.E. Biomineralization in the presence of calcium-binding phosphoprotein particles. J. Exp. Zoöl. 1986, 239, 207–220. [Google Scholar] [CrossRef]
- Inoue, H.; Ohira, T.; Ozaki, N.; Nagasawa, H. A novel calcium-binding peptide from the cuticle of the crayfish, Procambarus clarkii. Biochem. Biophys. Res. Commun. 2004, 318, 649–654. [Google Scholar] [CrossRef]
- Miyamoto, H.; Miyashita, T.; Okushima, M.; Nakano, S.; Morita, T.; Matsushiro, A. A carbonic anhydrase from the nacreous layer in oyster pearls. Proc. Natl. Acad. Sci. USA 1996, 93, 9657–9660. [Google Scholar] [CrossRef] [Green Version]
- Murayama, E.; Takagi, Y.; Ohira, T.; Davis, J.G.; Greene, M.I.; Nagasawa, H. Fish otolith contains a unique structural protein, otolin-1. Eur. J. Biochem. 2002, 269, 688–696. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M. Regulation of CaCO3 formation in coccolithophores. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2003, 136, 743–754. [Google Scholar] [CrossRef]
- Young, J.; Geisen, M.; Cros, L.; Kleijne, A.; Sprengel, C.; Probert, I.; Østergaard, J. A guide to extant coccolithophore taxonomy. J. Nannoplankton Res. 2003, 1, 1–125. [Google Scholar]
- Read, B.A.; Kegel, J.; Klute, M.J.; Kuo, A.; Lefebvre, S.C.; Maumus, F.; Grigoriev, I.V. Pan genome of the phytoplankton Emiliania underpins its global distribution. Nature 2013, 499, 209–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kegel, J.U.; John, U.; Valentin, K.; Frickenhaus, S. Genome Variations Associated with Viral Susceptibility and Calcification in Emiliania huxleyi. PLoS ONE 2013, 8, e80684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moheimani, N.R.; Borowitzka, M.A. Increased CO2 and the effect of pH on growth and calcification of Pleurochrysis carterae and Emiliania huxleyi (Haptophyta) in semicontinuous cultures. Appl. Microbiol. Biotechnol. 2011, 90, 1399–1407. [Google Scholar] [CrossRef]
- Westbroek, P.; Young, J.R.; Linschooten, K. Coccolith Production (Biomineralization) in the Marine Alga Emiliania huxleyi. J. Protozool. 1989, 36, 368–373. [Google Scholar] [CrossRef]
- Houdan, A.; Bonnard, A.; Fresnel, J.; Fouchard, S.; Billard, C.; Probert, I. Toxicity of coastal coccolithophores (Prymnesio-phyceae, Haptophyta). J. Plankton Res. 2004, 26, 875–883. [Google Scholar] [CrossRef]
- Hawkins, E.K.; Lee, J.J.; Fimiarz, D.K. Colony Formation and Sexual Morphogenesis in the Coccolithophore Pleurochrysis sp. (Haptophyta)1. J. Phycol. 2011, 47, 1344–1349. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y.; et al. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. GigaScience 2012, 1, 18. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Parra, G.; Bradnam, K.; Korf, I. CEGMA: A pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 2007, 23, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to Identify Repetitive Elements in Genomic Sequences. Curr. Protoc. Bioinform. 2009, 25, 4.10.1–4.10.14. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birney, E.; Clamp, M.; Durbin, R. GeneWise and Genomewise. Genome Res. 2004, 14, 988–995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanke, M.; Waack, S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics 2003, 19, ii215–ii225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burge, C.; Karlin, S. Prediction of complete gene structure in human genomic DNA. J. Mol. Biol. 1997, 268, 78–94. [Google Scholar] [CrossRef] [Green Version]
- Majoros, W.H.; Pertea, M.; Salzberg, S. TigrScan and GlimmerHMM: Two open source ab initio eukaryotic gene-finders. Bioinformatics 2004, 20, 2878–2879. [Google Scholar] [CrossRef]
- Alioto, T.; Blanco, E.; Parra, G.; Guigó, R. Using geneid to Identify Genes. Curr. Protoc. Bioinform. 2018, 64, e56. [Google Scholar] [CrossRef]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Pachter, L.; Salzberg, S.L. TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 2009, 25, 1105–1111. [Google Scholar] [CrossRef]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D.; Kelley, D.R.; Pachter, L. Differential gene and transcript expres-sion analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Regev, A. De novo transcript se-quence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith Jr, R.K.; Hannick, L.I.; White, O. Improving the Arabidop-sis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; E Allen, J.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annota-tion. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML: A program package for phylogenetic analysis by maximum likelihood. Bioinformatics 1997, 13, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Bie, T.; Cristianini, N.; DeMuth, J.P.; Hahn, M. CAFE: A computational tool for the study of gene family evolution. Bioinformatics 2006, 22, 1269–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Aris-Brosou, S.; Probert, I.; de Vargas, C. A Time line of the Environmental Genetics of the Haptophytes. Mol. Biol. Evol. 2010, 27, 161–176. [Google Scholar] [CrossRef] [Green Version]
- Szymanski, D.; Staiger, C.J. The Actin Cytoskeleton: Functional Arrays for Cytoplasmic Organization and Cell Shape Control. Plant Physiol. 2018, 176, 106–118. [Google Scholar] [CrossRef]
- Langer, G.; De Nooijer, L.J.; Oetjen, K. On the role of the cytoskeleton in coccolith morphogenesis: The effect of cytoskeleton inhibitors. J. Phycol. 2010, 46, 1252–1256. [Google Scholar] [CrossRef] [Green Version]
- Durak, G.M.; Brownlee, C.; Wheeler, G.L. The role of the cytoskeleton in biomineralisation in haptophyte algae. Sci. Rep. 2017, 7, 15409. [Google Scholar] [CrossRef] [Green Version]
- Suffrian, K.; Schulz, K.; Gutowska, M.A.; Riebesell, U.; Bleich, M. Cellular pH measurements in Emiliania huxleyi reveal pronounced membrane proton permeability. New Phytol. 2011, 190, 595–608. [Google Scholar] [CrossRef] [Green Version]
- Brownlee, C.; Taylor, A. Calcification in coccolithophores: A cellular perspective. Coccolithophores 2004, 31–49. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Total Base (Gb) | K-Mer | K-Mer Number | K-Mer Depth | Genome Size (Mb) | Revised Genome Size (Mb) | Heterozygous Ratio (%) | Repeat Ratio (%) |
|---|---|---|---|---|---|---|---|---|
| C. roscoffensis | 34.24 | 17 | 26,900,644,184 | 39 | 689.76 | 674.07 | 0.64 | 69.45 |
| Pair-End Libraries | Insert Size | Total Data (G) | Read Length (bp) | Sequence Coverage (X) |
|---|---|---|---|---|
| Illumina reads | 350 bp | 35.33 | 150 | 52.41 |
| Pacbio reads | 53.12 | 78.80 | ||
| 10X Genomics | 93.22 | 150 | 138.29 | |
| Total | 181.67 | 269.51 |
| Sample ID | Length | Number | ||
|---|---|---|---|---|
| Contig ** (bp) | Scaffold (bp) | Contig ** | Scaffold | |
| Total | 629,886,791 | 635,699,922 | 2167 | 769 |
| Max | 2,590,224 | 12,677,996 | ||
| Number ≥ 2000 | 2167 | 769 | ||
| N50 | 441,430 | 1,631,423 | 434 | 111 |
| N60 | 354,170 | 1,228,002 | 593 | 156 |
| N70 | 281,606 | 954,517 | 791 | 215 |
| N80 | 208,186 | 651,419 | 1053 | 296 |
| N90 | 141,820 | 391,115 | 1414 | 420 |
| Type | Repeat Size | % of Genome |
|---|---|---|
| Trf | 74,813,341 | 11.493833 |
| Repeatmasker | 327,015,645 | 50.240549 |
| Proteinmask | 67,002,054 | 10.293758 |
| Total | 381,019,300 | 58.537318 |
| Denovo + Repbase | TE Proteins | Combined TEs (All without Trf) | ||||
|---|---|---|---|---|---|---|
| Length (bp) | % in Genome | Length (bp) | % in Genome | Length (bp) | % in Genome | |
| DNA | 33,824,343 | 5.196551 | 4,008,971 | 0.615912 | 36,809,695 | 5.655201 |
| LINE | 7,142,576 | 1.097339 | 2,411,479 | 0.370484 | 8,374,515 | 1.286606 |
| SINE | 196,696 | 0.030219 | 0 | 0 | 196,696 | 0.030219 |
| LTR | 236,201,808 | 36.288504 | 60,676,043 | 9.321871 | 241,112,694 | 37.042981 |
| Other | 0 | 0 | 0 | 0 | 0 | 0 |
| Satellite | 3,083,747 | 0.473767 | 0 | 0 | 3,083,747 | 0.473767 |
| Simple_repeat | 25,608,316 | 3.934295 | 0 | 0 | 25,608,316 | 3.934295 |
| Unknown | 31,651,266 | 4.862694 | 0 | 0 | 31,651,266 | 4.862694 |
| Total | 327,015,645 | 50.240549 | 67,002,054 | 10.293758 | 331,759,778 | 50.969407 |
| Gene Set | Number | Average Gene Length (bp) | Average CDS Length (bp) | Average Exons Per Gene | Average Exon Length (bp) | Average Intron Length (bp) | |
|---|---|---|---|---|---|---|---|
| De novo | Augustus | 43,490 | 3611.96 | 1504.35 | 4.12 | 365.32 | 675.96 |
| GlimmerHMM | 313,490 | 1985.29 | 1123.67 | 3.85 | 292.11 | 302.67 | |
| SNAP | 102,913 | 1468.91 | 842.4 | 2.1 | 401.8 | 571.35 | |
| Geneid | 104,522 | 2507.4 | 1130.75 | 2.74 | 412.95 | 791.97 | |
| Genscan | 55,474 | 8837.72 | 2586.56 | 8.02 | 322.45 | 890.29 | |
| Homolog | Emiliania huxleyi | 21,246 | 1339.63 | 695.35 | 1.75 | 397.92 | 861.93 |
| Phaeodactylum tricornutum | 5755 | 1577.79 | 782.52 | 2.07 | 377.49 | 741.18 | |
| Chlamydomonas reinhardtii | 12,700 | 1608.62 | 938.93 | 1.92 | 489.67 | 729.92 | |
| Chlorella variabilis | 5117 | 1463.96 | 732.1 | 1.98 | 369.66 | 746.45 | |
| Volvox carteri | 13,333 | 922.29 | 609.61 | 1.47 | 413.34 | 658.52 | |
| Arabidopsis thaliana | 13,684 | 1312.01 | 892.26 | 1.47 | 609.02 | 902.56 | |
| RNA-seq | Cufflinks | 43,799 | 7548.43 | 2585.25 | 6.42 | 402.61 | 915.52 |
| PASA | 76,439 | 3568.24 | 1093.39 | 4.32 | 253.27 | 746.1 | |
| EVM | 47,323 | 3839.76 | 1523.32 | 4.34 | 351 | 693.55 | |
| PASA-update | 46,875 | 3848.09 | 1550.63 | 4.33 | 357.92 | 689.43 | |
| Final set | 23,341 | 5013.31 | 1596.61 | 5.75 | 277.68 | 719.32 | |
| Database | Annotated Num | Annotated Percent (%) | |
|---|---|---|---|
| NR | 16,841 | 72.2 | |
| Swiss-Prot | 11,919 | 51.1 | |
| KEGG | 11,807 | 50.6 | |
| InterPro | All | 23,179 | 99.3 |
| Pfam | 12,799 | 54.8 | |
| GO | 21,194 | 90.8 | |
| Annotated | 23,216 | 99.5 | |
| Total | 23,341 | - | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meng, R.; Zhang, L.; Zhou, C.; Liao, K.; Xiao, P.; Luo, Q.; Xu, J.; Cui, Y.; Hu, X.; Yan, X. Genome Sequence of Chrysotila roscoffensis, a Coccolithphore Contributed to Global Biogeochemical Cycles. Genes 2022, 13, 40. https://doi.org/10.3390/genes13010040
Meng R, Zhang L, Zhou C, Liao K, Xiao P, Luo Q, Xu J, Cui Y, Hu X, Yan X. Genome Sequence of Chrysotila roscoffensis, a Coccolithphore Contributed to Global Biogeochemical Cycles. Genes. 2022; 13(1):40. https://doi.org/10.3390/genes13010040
Chicago/Turabian StyleMeng, Ran, Lin Zhang, Chengxu Zhou, Kai Liao, Peng Xiao, Qijun Luo, Jilin Xu, Yanze Cui, Xiaodi Hu, and Xiaojun Yan. 2022. "Genome Sequence of Chrysotila roscoffensis, a Coccolithphore Contributed to Global Biogeochemical Cycles" Genes 13, no. 1: 40. https://doi.org/10.3390/genes13010040